Article Text

Abstract
Background: The most characteristic structural change evident in endobronchial biopsies in asthma, even in mild disease, is subepithelial collagen deposition within the lamina reticularis. This has been associated with progressive loss of lung function and the persistence of airway hyperresponsiveness, and has been linked to airway fibroblast proliferation. A potent fibroproliferative factor in bronchoalveolar lavage fluid in asthma is fibroblast growth factor-2 (FGF-2). FGF-2 is a member of a family of heparin binding growth factors that bind to heparan sulphate proteoglycans (HSPG), an important determinant of FGF-2 activity. This study compared the level of expression and distribution of FGF-2 in relation to HSPG in bronchial tissue from normal and asthmatic subjects.
Methods: The distribution of FGF-2 and HSPG in intact and cleaved forms in endobronchial biopsies from normal and asthmatic subjects was examined using an immunohistochemical approach. A novel ELISA based method was developed to detect solubilisation of FGF-2 following addition of heparin and heparitinase to bronchial tissue slices.
Results: Immunohistochemical analysis showed that FGF-2 was co-localised to HSPG in epithelial and endothelial basement membranes. Epithelial FGF-2, but not HSPG, was significantly more abundant in patients with mild asthma than in normal subjects. In vitro experiments indicated that FGF-2 was released from binding sites in the tissue by heparin and heparitinase I.
Conclusions: FGF-2 is bound by HSPG in bronchial tissue. The mast cell, through the release of heparin and endoglycosidase, may make a unique contribution to tissue remodelling in allergic asthma.
- fibroblast growth factor (FGF)-2
- heparin
- asthma
Statistics from Altmetric.com
A number of structural changes occur in the airway wall in asthma. The most characteristic is thickening of the subepithelial lamina reticularis which is observed in bronchial tissue even in patients with mild disease.1 This pathophysiological change—the result of deposition of interstitial collagens by increased numbers of myofibroblasts2—is likely to be directed by growth factors having fibroproliferative and profibrotic effects.
Fibroblast growth factor (FGF)-2 is a member of a family of heparin binding growth factors that affect the growth and differentiation of a large number of cell types.3 The FGFs are involved in morphogenesis, wound repair, inflammation, angiogenesis, and tumour growth and invasion,4 and require the glycosaminoglycan (GAG) side chains of heparan sulphate proteoglycans (HSPG) for high affinity binding to their specific receptors.5 Most cell surface HS is associated with two HSPG families, the syndecans and glypicans,6 whereas HS in the extracellular matrix is associated with perlecan and agrin.7 Perlecan is present in all basement membranes where it appears to function as a low affinity co-receptor for FGF-2.8 In addition to this role, binding to HS has been proposed to protect FGF-2 from proteolysis and to provide a reservoir of preformed growth factor that can be rapidly mobilised in response to appropriate stimuli.9 Recent evidence indicates an important role for bronchial fibroblasts in increased proteoglycan deposition associated with bronchial hyperresponsiveness in the asthmatic airway.10
FGF-2 is ubiquitous in normal human tissues where it is found in association with endothelial basement membranes.11 At this site it appears to be sequestered in a stable but inactive form requiring release for biological activity.12 A number of mechanisms for the release of FGF-2 from extracellular matrices have been identified. These include proteolytic cleavage of HSPG core proteins,13,14 the actions of GAG degrading enzymes,12,15 and the ability of heparin to elute FGF-2 from HSPG binding sites.12,16 HSPGs are shed efficiently from cell surfaces by the action of exogenous enzymes such as plasmin and thrombin, and by endogenous matrix metalloproteinases (MMPs).6
Levels of FGF-2, a potent fibroblast mitogen, are increased in bronchoalveolar lavage (BAL) fluid in patients with mild asthma and are further increased in the airways of individuals with allergic asthma after endobronchial allergen challenge.17 In view of the potentially important role for FGF-2 in the pathogenesis of tissue remodelling in asthma, we have used an immunohistochemical approach to compare the distribution of FGF-2 and HSPG in bronchial tissue from patients with mild asthma and normal control subjects. Additionally, we have developed a novel assay to detect FGF-2 release from bronchial tissue ex vivo in response to heparin and the HS degrading enzyme heparitinase I.
METHODS
Subjects
Two weeks before bronchoscopy subjects attended for skin prick testing and for measurement of prebronchodilator forced expiratory volume in 1 second (FEV1) and airway responsiveness. Histamine inhalation challenge to determine the provocative concentration required to produce a 20% fall in FEV1 (PC20) was undertaken as previously described18 by a modification of the method of Chai et al.19 For immunolocalisation studies we recruited seven asthmatic subjects (one male) aged 33.0 (3.4) years with FEV1 95.0 (3.8)% predicted and PC20 histamine 0.83 (0.2–10.1) mg/ml, and six non-asthmatic control subjects (five male) aged 25.7 (2.5) years with FEV1 100.7 (3.6)% predicted. Six of the asthmatics and four of the control subjects were atopic. For tissue slice experiments we recruited separate groups of five atopic asthmatic subjects (four male) aged 39.3 (5.7) years with FEV1 87.1 (2.7)% predicted and PC20 histamine 2.23 (0.34–14.5) mg/ml and four atopic non-asthmatic control subjects (all male) aged 26.5 (3.8) years with FEV1 100.3 (2.9)% predicted. Asthmatic subjects were mildly symptomatic and were receiving inhaled salbutamol as required as their only treatment. All non-asthmatic control subjects had histamine PC20 measurements outside the asthmatic range (>32 mg/ml).
The study was approved by the combined Southampton Hospitals and University Research ethics committee and all subjects gave written informed consent.
Bronchoscopy and tissue processing
Fibreoptic bronchoscopy was performed in accordance with our previously published protocol18 and the recommendations of the National Heart, Lung, and Blood Institute.20 Bronchial biopsies (1–2 mm diameter) were obtained from third or fourth generation airway carinae and immediately either fixed for immunohistochemistry in ice cold acetone containing the protease inhibitors iodoacetamide (20 mM) and phenyl methyl sulphonyl fluoride (2 mM) or snap frozen in liquid nitrogen for tissue slice experiments.
Immunohistochemistry
Specimens for immunohistochemistry were processed into glycolmethacrylate (GMA) resin as previously described.21 Thin (2 μm) sections were cut from GMA embedded tissue, floated on 0.2% (v/v) ammonia in double distilled water, and collected onto poly-l-lysine coated glass slides. Sections were dried for 1 hour at room temperature and wrapped in aluminium foil for storage at −20°C.
Immunohistochemical staining was carried out using our published protocol.21 Endogenous peroxidase activity was inhibited by applying a solution of 0.3% hydrogen peroxide in 0.1% sodium azide for 30 minutes, and non-specific binding was blocked by applying a solution of 10% fetal bovine serum plus 1% BSA in Dulbecco’s modified Eagle’s medium (GibCo, Paisley, UK). All washes were in 0.05 M Tris buffered saline, pH 7.6 (TBS). The following primary antibodies were used: 1:25 dilution of mouse IgG1 monoclonal anti-human FGF-2 (Oncogene, Boston, MA, USA), 1:50 dilution of mouse IgM monoclonal anti-HS (10E4) which recognises intact HS, and 1:50 dilution of mouse IgG2b monoclonal anti-HS (3G10) which recognises the terminal glucuronate residue following heparitinase digestion of HSPG (Seikagaku Corp, Tokyo, Japan). A detailed characterisation of the properties of these two anti-HS antibodies has been reported previously.22 Control sections were similarly treated, but the primary antibody was either omitted or replaced with an isotype matched control antibody (Sigma Chemical Co, Poole, UK) at the same concentration as the test antibody. Biotinylated rabbit anti-mouse F(ab′)2 fragments (Dako, High Wycombe, Bucks, UK) were applied at 1:300 dilution for 2 hours. Streptavidin-biotin-peroxidase complex (Dako) was applied for 2 hours to amplify detection and aminoethyl carbazole (0.04%)/hydrogen peroxide (0.045%) solution applied as the chromogenic substrate for 30 minutes at 37°C. The slides were then rinsed in TBS, washed in cold running tap water, counterstained with Mayer’s haematoxylin and blued in cold running tap water. Finally, sections were covered with Crystalmount (Biogenesis, Poole, UK), dried at 80°C and mounted in DPX (BDH Ltd, Poole, UK).
The distribution and relative expression of FGF-2 and HS were assessed using computerised image analysis as previously described.23 Blood vessels were identified by immunostaining (as above) with a 1:80 dilution of a mouse IgG1 monoclonal antibody to the endothelial cell antigen EN4 (Monosan, Uden, NL) and counting the whole area of the section using a light microscope at a magnification of ×40. For FGF-2, epithelial staining was analysed as the percentage total area of epithelium plus basement membrane and endothelial staining was represented as the percentage total number of blood vessels staining positively. For both forms of HS, staining was analysed as the percentage total length of epithelial basement membrane and percentage total number of blood vessels staining positively.
Release of FGF-2 from tissue slices
To measure the basal and stimulated release of FGF-2 from bronchial tissue, a novel one step method for the rapid capture, detection, and quantitation of the growth factor was developed. Tissue slices (20 μm cryosections) were cut from frozen biopsies embedded in OCT mounting medium (Tissue Tek II, Miles Laboratories Inc, Naperville, USA) and placed in 200 μl cold RPMI medium (GibCo) in the wells of a 96-well microtitre plate precoated with anti-FGF-2 (R&D Systems, Oxford, UK). Plates were warmed to room temperature before adding stimuli. Heparin from pig intestinal mucosa with molecular weight 13 500–15 000 (Calbiochem, Nottingham, UK), the HS degrading endoglycosidase heparitinase I (Seikagaku Corp), MMP-3 (Biogenesis), or streptokinase (Sigma Chemical Co) were added at the final concentrations and incubated for 10, 15, 20, 40, and 60 minutes at 37°C. FGF-2 standards were included at each time point because the sensitivity of the ELISA is reduced at time points less than 60 minutes. Incubations were carried out in duplicate using non-adjacent sections. To test for protease dependent release of FGF-2, 100 μg/ml α2-macroglobulin was added in some experiments. Following incubation, tissue and medium were removed, the wells washed, and FGF-2 that had been released and captured by the primary antibody was quantitated by completing the subsequent ELISA steps according to the manufacturer’s instructions.
Statistical analysis
Clinical data for age and FEV1 % predicted were expressed as mean (SE) and for PC20 as geometric mean (range). Immunohistochemical data were expressed as median (range) and compared using the Mann-Whitney U test and the Wilcoxon test for unpaired and paired data, respectively. FGF-2 concentrations in culture supernatants were expressed as mean (SE). Release of FGF-2, expressed as percentage change from baseline, was analysed using a repeated measures ANOVA model with a post hoc Fisher’s PLSD test for concentration-response experiments, an unpaired t test to compare tissue from asthmatic and non-asthmatic subjects, and a paired t test to study the effect of α2-macroglobulin. Analysis was performed using StatView 5.01 for Macintosh computers (Abacus Concepts, Berkeley, CA, USA). A significance level of 5% was accepted.
RESULTS
Immunolocalisation of FGF-2 and HS in bronchial tissue
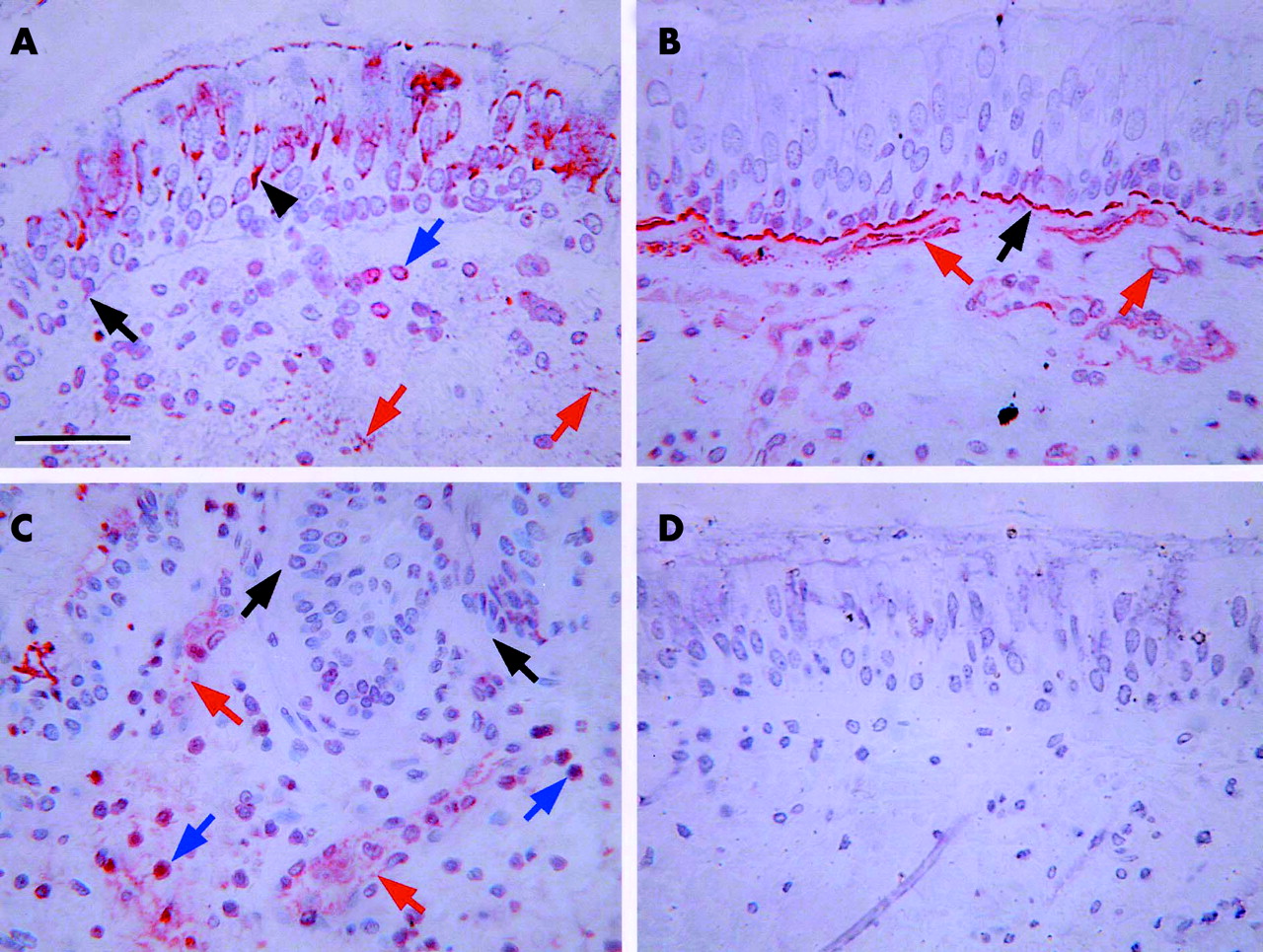
Intracellular and extracellular FGF-2 immunoreactivity was detected in bronchial tissue from control (fig 1A⇓) and asthmatic subjects. Intracellular FGF-2 was observed within bronchial epithelial cells and in cells within the subepithelial region. Extracellular FGF-2 was seen in the pericellular matrix of endothelial cells and in the epithelial basement membrane. The intact form of HS was detected in a linear distribution on epithelial and endothelial basement membranes where it was found in abundance in both control (fig 1B⇓) and asthmatic tissue. Intracellular intact HS was detected only in endothelial cells and could be detected at both lumenal and basal aspects of these cells. The cleaved form of HS was also found predominantly in endothelial basement membranes and, notably in the asthmatics, in many large mononuclear cells in the tissue (fig 1C⇓). However, these same cell types did not stain for intact HS. No immunoreactivity was detected if the primary antibody was replaced with an isotype matched control mouse IgG1 (fig 1D⇓) or if the primary antibody was omitted (not shown).
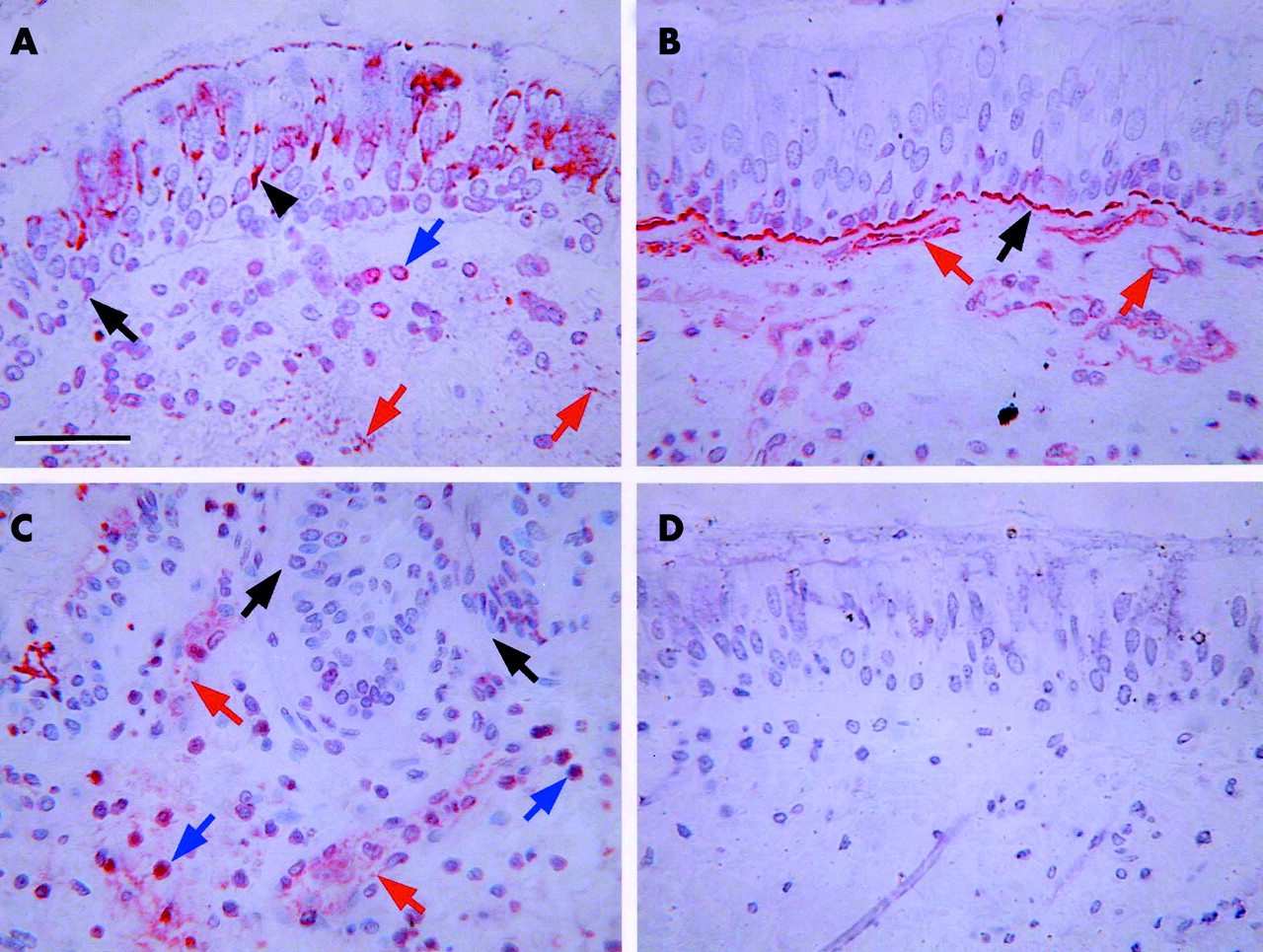
Representative immunohistochemical staining patterns. (A) Anti-FGF-2 in tissue from a control subject. (B) Antibody 10E4 (to identify intact heparan sulphate (HS)) in tissue from a control subject. (C) Antibody 3G10 (to identify cleaved HS) in tissue from an asthmatic subject. (D) Control section stained with an IgG1 isotype control antibody. Black arrows indicate epithelial basement membrane, black arrowheads indicate epithelium, red arrows indicate endothelium, and blue arrows indicate positively staining cells in the submucosa. Scale bar = 50 μm.
Quantitative analysis indicated a greater area of epithelial FGF-2 immunostaining in asthmatic tissue than in control tissue (median 8.7% v 5.3%, p<0.05; fig 2⇓). However, there was no significant difference in the proportion of blood vessels staining positively for FGF-2 between these two subject groups (median 36% v 29.5%, p = 0.89). The cleaved form of HS was significantly less abundant than the intact form at the endothelial level and in the normal epithelial basement membrane (table 1⇓). The proportion of epithelial basement membrane and the proportion of blood vessels staining positively for both intact and cleaved forms of HS did not differ significantly between the two subject groups (table 1⇓).
Quantitation of immunohistochemical staining for intact and cleaved forms of heparan sulphate (HS) in bronchial tissue from seven asthmatic subjects and six non-asthmatic control subjects
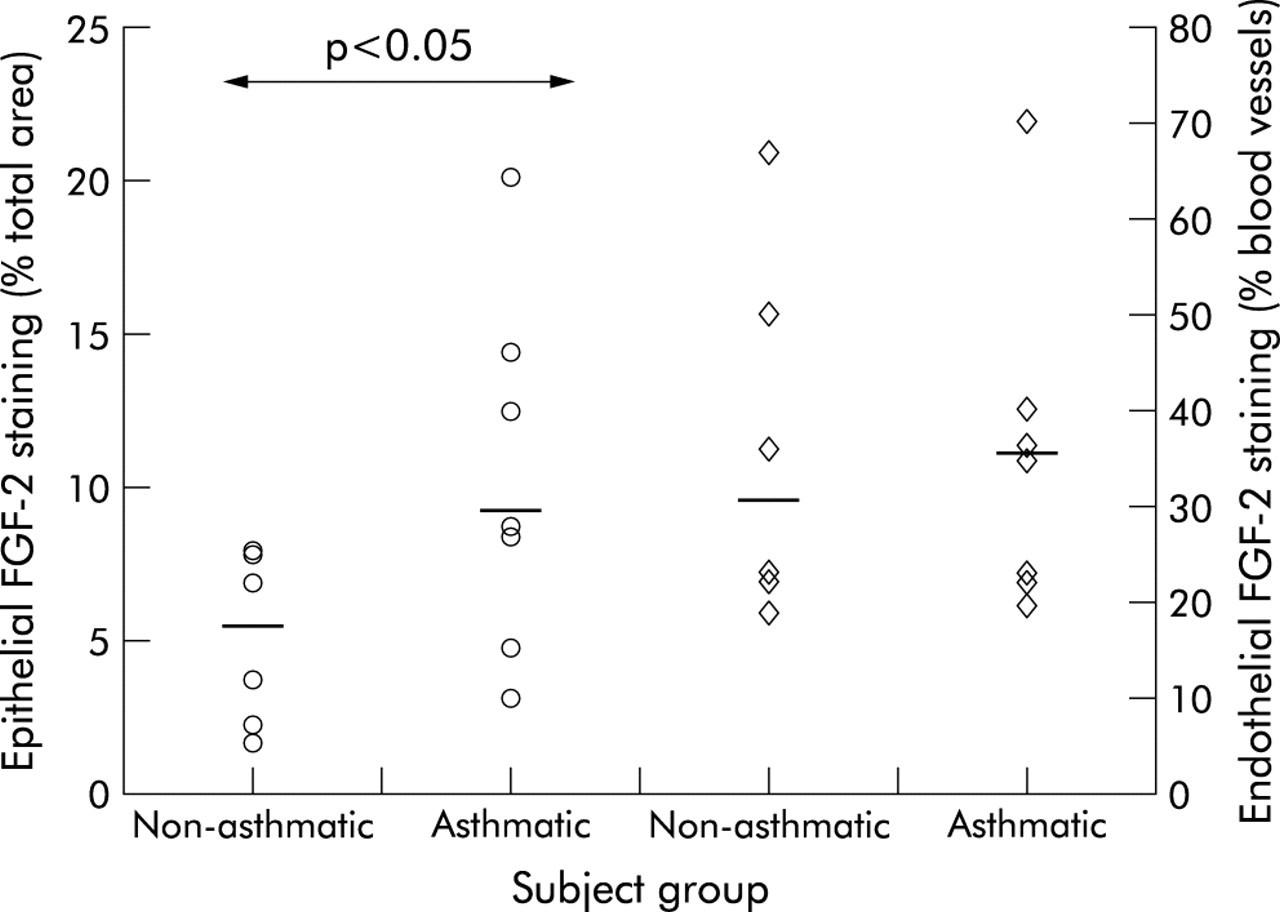
Quantitation of FGF-2 immunostaining in the bronchial epithelium (circles) and vascular endothelium (diamonds) in asthmatic and non-asthmatic subjects. Horizontal bars represent median values. Between group comparisons were performed using the Mann-Whitney U test.
Release of FGF-2 from bronchial tissue
In preliminary experiments FGF-2 was undetectable in the supernatants of bronchial tissue slices incubated in tissue culture plastic ware with or without stimuli. We suspected that this was due to rebinding of FGF-2 to tissue sites. Therefore, to prevent rebinding, tissue slices were incubated directly in the wells of ELISA plates in which the specific high affinity primary antibody used to coat the wells would effectively compete with tissue binding sites and capture the released growth factor. Using this assay we investigated the effect of substances that might influence the binding of FGF-2 to HS side chains.
Heparin
No significant effect was observed at incubation times of 10, 15, 20, or 40 minutes at any concentration of heparin (n = 4, results not shown). At an incubation time of 60 minutes (n = 6) there was a concentration-dependent increase in FGF-2 release (fig 3A⇓). This effect was of borderline significance (p = 0.055) at a heparin concentration of 10 μg/ml. At 100 μg/ml heparin there was a highly significant (p = 0.0001) effect with FGF-2 levels increased to a mean of 217% (95% CI 140 to 293) of baseline—that is, from 29.1 (6.4) pg/ml to 58.7 (12.5) pg/ml. There was no further increase in FGF-2 release at a heparin concentration of 200 μg/ml (52.7 (17.1) pg/ml, n = 3).

FGF-2 release from bronchial tissue slices (n = 6 except heparin 200 μg/ml where n = 3). (A) Concentration-response to heparin at 60 minute incubation time. (B) Concentration-response to heparitinase I at 60 minute incubation time. Bars represent the mean and error bars the SE. Analysis was performed using a repeated measures ANOVA with a post hoc Fisher’s PLSD test for multiple comparisons. Asterisks indicate a significant increase above basal release.
Additional experiments were performed at optimal conditions (heparin 100 μg/ml for 60 minutes) to compare tissue slices from atopic asthmatic donors (n = 5) and atopic non-asthmatic donors (n = 4) and to study the effect of the protease inhibitor α2-macroglobulin (n = 3). There was a trend for a greater response in tissue from the asthmatic group (mean 237% v 171% of baseline), but this difference did not reach statistical significance (p = 0.11). Inclusion of α2-macroglobulin did not significantly alter unstimulated release of FGF-2 (60.8 (14.4) pg/ml v 44.9 (9.5) pg/ml, p = 0.44) and did not inhibit release induced by heparin (fig 4⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
FGF-2 release was measured in response to heparin and heparitinase I in the absence and presence of α2-macroglobulin (100 μg/ml) and compared with basal release which was designated as 100% (n = 3). Bars represent the mean and error bars the SE.
Heparitinase I
No significant effect of heparitinase I at any concentration was observed at incubation times of 10, 15, 20, or 40 minutes (n = 4, results not shown). At an incubation time of 60 minutes (n = 6) heparitinase I concentrations of 2.5 mU/ml and 12.5 mU/ml were without effect but 25 mU/ml heparitinase I significantly (p = 0.0048) increased FGF-2 levels to a mean of 249% (95% CI 102 to 397) of baseline—that is, from 37.4 (13.4) pg/ml to 70.7 (18.21) pg/ml (fig 3B⇑).
Additional experiments, as described above for heparin, were performed with heparitinase 25 mU/ml for 60 minutes. There was no significant difference in the magnitude of the response between tissue from asthmatic and non-asthmatic subjects (mean 237% v 195% baseline, p = 0.64). The effect of heparitinase I was not inhibited by the protease inhibitor α2-macroglobulin at 100 μg/ml (fig 4⇑).
Matrix metalloproteinase-3 (MMP-3, stromelysin) and streptokinase
For comparison, the effects of enzymes that might lead to cleavage of proteoglycan core proteins—either directly or indirectly via activation of other proteases—were investigated. Incubation of tissue slices with MMP-3 (130 ng/ml) or streptokinase (500 IU/ml) for 60 minutes did not increase the release of FGF-2.
DISCUSSION
This study describes the localisation and relative abundance of immunoreactive FGF-2 and HS in bronchial tissue from subjects with mild asthma and healthy control subjects. We have shown for the first time that epithelial FGF-2 immunostaining is increased in asthmatic tissue. HS was present in epithelial basement membranes and at vascular sites, but quantitative analysis revealed no detectable difference between the subject groups at these locations. We have also described a novel method to detect release of FGF-2 from bronchial tissue slices and found that heparin and heparitinase I induce FGF-2 release in a time and concentration dependent manner.
We have previously identified higher basal levels of FGF-2 in BAL fluid in patients with atopic asthma.17 In principle, this could represent increased generation and release or, alternatively, decreased catabolism. The higher baseline levels in BAL fluid may reflect the increased expression of FGF-2 by epithelial cells in asthma that we have shown in this study. A recent report has also indicated increased FGF-2 expression in asthmatic subjects by cells in the submucosa which were identified in order of abundance as CD34+ cells > eosinophils > macrophages > T cells > mast cells.24 FGF-2 is a mitogen for many cells including fibroblasts and endothelial cells—cell types that express CD34—and these cells may therefore be both a source and a target of FGF-2 activity.
The co-localisation of FGF-2 and HS in bronchial tissue suggests that FGF-2 is largely bound in an inactive form on the endothelium and in epithelial basement membranes, providing an extracellular reservoir of the growth factor. FGF-2 binds to the HS side chains of HSPG, the major pericellular HSPGs being perlecan in the basement membrane and the syndecans and glypicans of the cell surface.25 Because cleavage of HS side chains may indicate local release of bound growth factor, we studied the expression of HS using two specific antibodies—one which recognises intact HS chains and one which recognises an epitope generated by heparitinase digestion of HSPG.22 Intact HS was the predominant form in both normal and asthmatic tissue. We had anticipated that cleaved HS might be more abundant in asthmatic tissue but, in fact, we could find no significant difference between the subject groups in the distribution or abundance of either form of HS at epithelial and endothelial sites. However, the variability of measurements and the sample size may have limited the ability of the study to detect such a difference. In contrast, a recent report indicates increased expression of perlecan, biglycan, and small HSPGs by fibroblasts from subjects with mild asthma.10 The mononuclear cells that stained for cleaved but not intact HS in asthmatic tissue in the present study may have been monocytes/macrophages—cells that express HS on maturation in tissues but not in the blood26 and constitutively release HS degrading activity on contact with tissue matrices.27
We previously speculated that the significant increase in FGF-2 in the BAL fluid following endobronchial allergen challenge, which occurs within 10 minutes and is therefore too early to reflect de novo synthesis, may have reflected the release of preformed FGF-2 from storage sites in bronchial tissue in response to enzymatic cleavage of HS or proteoglycan displacement.17 Because the rapid in vivo release of FGF-2 occurred in temporal association with mast cell degranulation, we investigated the effects of heparin and heparitinase I on FGF-2 release using an ex vivo tissue slice model.
Heparin has an important impact on FGF-2 activity. A recent study has indicated dual functions of heparin in the regulation of this activity: cell surface bound heparin potentiates FGF-2 receptor interactions whereas soluble heparin binds FGF-2 in solution and inhibits its interaction with specific cell surface receptors.28 Heparin may therefore either stimulate or inhibit FGF-2 receptor binding and bioactivity, the net effect being dependent on the concentration of heparin, the number of HS sites on the target cell, and the affinity and number of binding sites for heparin on the target cell.28
In the present study we have shown that heparin stimulates the release of FGF-2 from bronchial tissue slices. The concentration of heparin required (100 μg/ml) was higher than that reported to displace FGF-2 from isolated bovine corneal basement membrane (10 μg/ml)12 and from the subendothelial extracellular matrix produced by cultured endothelial cells (5 μg/ml),16 perhaps reflecting the greater number of non-specific heparin binding sites in whole tissue. However, the effective concentration of heparin was of the order of that previously estimated to be released by sensitised human lung tissue fragments after allergen challenge (mean approximately 19.3 μg/g tissue),29 although concentrations of heparin achieved locally in the immediate vicinity of mast cells after release are presumably much higher. Although we previously detected FGF-2 release into BAL fluid within 10 minutes of allergen exposure,17 release from tissue slices in the present study was not evident at incubation times shorter than 60 minutes. The reason for this difference between the in vivo and in vitro kinetics of FGF-2 release is unclear.
Enzymes that cleave HSPGs—either their core proteins or GAG side chains—are likely to influence the form and function of FGF-2. For example, the bacterial endoglycosidase heparitinase I has been shown to reduce FGF-2 binding to fibroblasts and to abrogate FGF-2 induced proliferation through cleavage of the cell surface HSPG syndecan-4.30 Kato et al31 recently identified a novel pathway for the regulation of FGF-2 activity involving another cell surface HSPG syndecan-1. Under physiological conditions the soluble syndecan-1 ectodomain potently inhibits heparin induced FGF-2 mitogenicity. However, partial degradation of the GAG chains by bacterial heparitinase or platelet heparanase, as may occur at sites of inflammation and tissue injury, converts syndecan-1 to an FGF-2 activator by generating stimulatory oligosaccharide sequences.31 The activity of FGF-2 at cell surfaces may therefore be regulated by the shedding and limited degradation of HSPGs. In the present study, extracellular FGF-2 was immunolocalised to the lumenal and basal surfaces of endothelial cells and to the epithelial basement membrane. Release of FGF-2 following incubation of bronchial tissue with heparitinase I confirms that the growth factor was bound to HS side chains of HSPG and indicates a potential mechanism for FGF-2 release at sites of allergic inflammation. Plasminogen activators, acting directly or as activators of a number of MMPs, have been implicated in the release of FGF-2 from endothelial cells.13 At the concentrations tested, however, we were unable to demonstrate any effect of the plasminogen activator streptokinase or of the proteoglycanase MMP-3 on FGF-2 release. The same nanomolar concentration of MMP-3 was previously shown in an in vitro model to induce damage and loss of GAGs from gut mucosal tissue, an effect that could be inhibited with α2-macroglobulin.32 The lack of effect of α2-macroglobulin at the same concentration in our study suggests that heparin and heparitinase I were acting directly on the GAG side chains of tissue HSPG.
The studies reported here link an aspect of airway inflammation in asthma—namely, mast cell activation—with an aspect relevant to the remodelling process in this disease—namely, FGF-2 release. Activation of FGF-2 by release from sites of encryption is likely to contribute to the structural changes seen in the asthmatic airway, including fibroblast proliferation and angiogenesis. The modulation of the activity of this and other growth factors—such as transforming growth factor β (TGF-β) and platelet-derived growth factor (PDGF)—by proteoglycan binding represents an important post-translational regulatory process and indicates that mechanisms relevant to the chronic structural airway changes can occur in parallel with the inflammatory process. A better understanding of other factors involved in post-translational regulation of growth factor bioavailability and the influence of airway anti-inflammatory therapy on these processes is thus warranted.
Acknowledgments
The authors acknowledge the help of Miss Helen Rigden in completing the immunohistochemical analysis and Dr Anton Page for preparing the colour images. This work was supported by the Medical Research Council of Great Britain (grant number G8604034).