Article Text

Abstract
Background Mechanisms that facilitate early infection and inflammation in cystic fibrosis (CF) are unclear. We previously demonstrated that children with CF and parental-reported secondhand smoke exposure (SHSe) have increased susceptibility to bacterial infections. SHSe hinders arachidonic acid (AA) metabolites that mediate immune function in patients without CF, and may influence CF immune dysfunction. We aimed to define SHSe’s impact on inflammation mediators and infection in children with CF.
Methods Seventy-seven children with CF <10 years of age (35 infants <1 year; 42 children 1–10 years) were enrolled and hair nicotine concentrations measured as an objective surrogate of SHSe. AA signalling by serum and macrophage lipidomics, inflammation using blood transcriptional profiles and in vitro macrophage responses to bacterial infection after SHSe were assessed.
Results Hair nicotine concentrations were elevated in 63% of patients. Of the AA metabolites measured by plasma lipidomics, prostaglandin D2 (PGD2) concentrations were decreased in children with CF exposed to SHSe, and associated with more frequent hospitalisations (p=0.007) and worsened weight z scores (p=0.008). Children with CF exposed to SHSe demonstrated decreased expression of the prostaglandin genes PTGES3 and PTGR2 and overexpression of inflammatory pathways. These findings were confirmed using an in vitro model, where SHSe was associated with a dose-dependent decrease in PGD2 and increased methicillin-resistant Staphylococcus aureus survival in human CF macrophages.
Conclusions Infants and young children with CF and SHSe have altered AA metabolism and dysregulated inflammatory gene expression resulting in impaired bacterial clearance. Our findings identified potential therapeutic targets to halt early disease progression associated with SHSe in the young population with CF.
- cystic fibrosis
- macrophage biology
- tobacco and the lung
- paediatric lung disaese
Statistics from Altmetric.com
Key messages
What is the key question?
What are the mechanisms associated with increased inflammation and infection due to secondhand smoke exposure in infants and young children with cystic fibrosis (CF)?
What is the bottom line?
We showed that secondhand smoke exposure assessed objectively is highly prevalent in young children with CF and results in alterations in arachidonic acid metabolism and inflammatory gene expression, which in turns impairs bacterial clearance.
Why read on?
Our findings highlight the important role secondhand smoke exposure plays in modifying early outcomes in CF and provide details for future studies and interventions.
Introduction
Despite recent advances in treatment modalities, cystic fibrosis (CF) remains the most common, life-limiting, genetic disease among European descendants. Disease progression begins early in life in these patients due to cystic fibrosis transmembrane conductance regulator (CFTR) dysfunction. Early disease is influenced by both host and environmental factors. Secondhand smoke exposure (SHSe) is a common factor known to increase asthma severity and respiratory infections in children without CF,1 and may disproportionately affect infants and young children with CF.2 3
While inflammation is thought to play a role in early disease progression,4–7 the contribution of environmental factors such as SHSe on infection and inflammation is poorly understood in children with CF. Our previous studies suggested that SHSe was highly prevalent (44%) in young children with CF and it was associated with an increased incidence of chronic bacterial infections and worsened clinical outcomes.3 Alterations in arachidonic acid (AA) metabolism and its subsequent impact on host immunity have been demonstrated in smokers without CF and adults with asthma.8 9 Dysregulated AA metabolism has also been shown during CF exacerbations and periods of poor growth, but it has not been linked to SHSe in children with CF.10 11 Although longitudinal studies have shed light on clinical,10 12–14 microbiological15 and environmental16 factors that impact long-term lung disease, modifiable factors such as SHSe and their interaction with immunological, microbiological or AA signalling in infants and young children with CF are relatively unknown.
We hypothesised that SHSe impairs CF macrophage function and bacterial killing through altered AA metabolism and an exaggerated inflammatory response. To test this hypothesis, we studied AA signalling, inflammation and macrophage function in young children with CF and objectively measured SHSe to identify potential areas for therapeutic intervention in this vulnerable population.
Methods
Subjects
Children aged 4 months–10 years with CF were recruited from outpatient CF clinics, and stratified according to age (infants<1 year, and older children). The diagnosis of CF was defined by two disease-causing mutations or a sweat chloride test ≥60 mmol/L. All patients except one infant had at least one copy of the Phe508del mutation. A parent or guardian of the child participant provided informed consent on their behalf. Healthy, non-CF age-matched controls were recruited from primary care clinics.
Clinical measures
We collected for each patient at clinic visits: clinical information using a standardised clinical questionnaire, hair to measure nicotine concentrations, serum for lipidomics, whole blood for transcriptional profiling and oropharyngeal swabs or sputum for bacterial growth assessment. Patients also underwent pulmonary function testing. Lung function was measured as FEV in 0.5 s (FEV0.5) and 1 s (FEV1) for infants and children, respectively. Percent predicted measurements and z scores for forced vital FEV0.5 and FEV1 were derived from reference equations.17 18 Growth was quantified as height and weight z scores by CF dieticians trained in anthropometric measurements using WHO standardised growth charts. Z scores represent standard measures of weight in the CF population and were selected based on prior studies documenting their utility as a marker of nutritional status in CF.19 20 Hospitalisations for pulmonary exacerbations were recorded for the 6 months prior to study inclusion. Pulmonary exacerbations were verified according to a previously published definition.21 Determination of bacterial growth was performed by routine oropharyngeal culture or sputum collection at each study visit. If a subject underwent clinical bronchoscopy during the study period, bronchoalveolar lavage results were also recorded.
Blood samples from additional children and adults with and without CF were obtained for in vitro studies as well as from age, gender and race-matched healthy children for gene expression analyses.
Secondhand smoke exposure
Hair nicotine concentrations were assessed and considered as the primary objective measure of SHSe. Hair nicotine provides a long-term, stable measure of SHSe as nicotine is integrated into the growing hair shaft over multiple months.22 Hair was obtained following a standardised protocol (see online data supplement).23 Samples were processed by reverse-phase high-performance liquid chromatography with electrochemical detection as described.22 Lower limits of detection (0.1 ng/mg) may distinguish poorly SHSe from no exposure,24 therefore we selected a more conservative cut-off (1.0 ng/mg) to dichotomize groups into (+) and (−) SHSe.
Supplemental material
Biomarker measurements and study design
Our primary outcome was to assess the effects of SHSe on the activation or suppression of AA pathways in children with CF. Additionally, we assessed as secondary outcomes the impact of SHSe and AA metabolites on: a) clinical outcomes, b) inflammatory pathways by transcriptome analysis and c) bacterial clearance and macrophage function. To this end, serum and blood samples were collected for each individual patient, and detailed methods are described in the online data supplement. We applied the following analytical strategies to the original cohort composed of 57 children (15 infants and 42 children aged 1–10 years) with AA data validated in an additional 20 infants with CF: (1) AA and immune/inflammatory markers were determined by serum lipidomics and whole blood transcriptional profiles, respectively; (2) SHSe, AA and inflammatory/immune profiles were correlated with clinical outcomes that included lung function, growth, pulmonary exacerbations and bacterial growth; (3) finally, to confirm the findings derived from lipidomics and transcriptional profiling, we assessed in vitro monocyte-derived macrophage (MDM) function after exposure to cigarette smoke extract (CSE) as described in the online data supplement.
Statistical analysis
CF group demographic characteristics were compared using one-way analysis of variance. Unpaired t-test or Mann-Whitney U test were used to compare blood and macrophage AA metabolites or hair nicotine concentrations according to data distribution. Two sample t-test or Mann–Whitney U test was used for comparisons of independent in vitro experiments. Wilcoxon rank-sum test was used to compare AA metabolites with need for hospitalisation. Pearson’s correlation coefficient was computed for correlations between AA metabolites and weight z score or FEV1. For transcriptional profile analyses, we used stringent statistical filters and established protocols as described (see online data supplement for full details).25 26 Data analysis was performed in Stata/MP V.13.1, R software and GraphPad Prism V.7.0, with a two-tail p<0.05 considered to be statistically significant.
Results
Cohort characteristics and SHSe
Patient demographics are listed in table 1 for the CF cohort and additional non-CF controls used in transcriptomics and in vitro studies. This original cohort of 57 infants and children with CF and 20 non-CF controls was enrolled from a single institution (Nationwide Children’s Hospital (NCH)). Demographic characteristics of a validation cohort of 20 additional infants with CF used in confirmatory AA studies (from NCH and Riley Children’s Hospital (RCH)) are also listed in table 1. Overall, the cohort demographics and clinical characteristics were similar between infants and children with CF. Most patients had pancreatic insufficiency and had at least one copy of the Phe508del mutation. Fifty-five per cent of children vs 20%–33% of infants had a history of infection with Pseudomonas aeruginosa (p=0.026), whereas history of methicillin-resistant Staphylococcus aureus (MRSA) infection was 31% for children vs 10%–20% for infants (p=0.18). On the other hand, oropharyngeal cultures obtained at enrolment in all study patients revealed that P. aeruginosa and Stenotrophomonas maltophilia were significantly more frequent in children with CF exposed to SHSe compared with those not exposed to SHSe (P. aeruginosa, 54.8% vs 23.1%, p=0.05) (S. maltophilia 45.2% vs 7.7%, p=0.016). MRSA positivity rate was not different between groups, but the overall rate of detection was low (28%).
Cohort demographics
The distribution of nicotine concentrations in infants and children with CF included in the original cohort are shown in figure 1 and median values reported in table 1. For figure 1, children were grouped by age in infants (<1 year), preschool/daycare age (1–5 years) and school age (≥6–10 years) due to the potential for less exposure to nicotine once in school. Nicotine concentrations were elevated in 63% of all patients with CF, with a median concentration of 2.3 (0.1–9.1) ng/mg hair. Higher nicotine concentrations were detected in infants with CF compared with children (24.9 (0.8–39.4) vs 1.7 (0.1–5.5) ng/mg, respectively), and inversely correlated with age (r=−0.33, p<0.011). There was no difference in nicotine concentrations between school age children and children with CF aged 1–5 years. As a reference, active smokers have been reported to have median hair nicotine concentrations of 16.2 ng/mg hair.24

Infants with cystic fibrosis (CF) have disproportionately high secondhand smoke exposure. CF hair nicotine concentrations were determined by high-performance liquid chromatography-tandem mass spectrometry and plotted by age grouping (infants<1 year, children 1–5 years, children≥5 years). P=0.006 for infants vs children 1–5 years, p=0.002 for infants vs children≥5 years, via t-tests. Analysis of variance p=0.002. Nicotine concentrations were inversely correlated with age (r=−0.33, p=0.011, Pearson’s correlation coefficient), n=57. As a reference, active smokers have been reported to have median hair nicotine concentrations of 16.2 ng/mg hair.
To examine specific changes in AA metabolites in infants and children with CF exposed to SHSe and not exposed to SHSe, lipidomics profiles were analysed in serum samples via high-performance liquid chromatography-tandem mass spectrometry in the original NCH cohort. Both infants and children with CF in whom hair nicotine concentrations (as a surrogate of SHSe) were above the cut-off had a significant decrease in a non-dose-dependent manner in the following AA metabolites: prostaglandin D2 (PGD2), leukotriene C4, 8S-hydroxy-5Z,9E,11Z,14Z-eicosatetraenoic acid (8(S)-HETE), 9S-hydroxy-5Z,7E,11Z,14Z-eicosatetraenoic acid (9(S)-HETE), 12(S)-hydroperoxy tetraenoic eicosatetraenoic acid (12(S)-HETE) and 14(15)-epoxy-5Z,8Z,11Z-eicosatrienoic acid (14(15)-EET) (pathway—figure 2A, concentrations—online supplement table E1).

Secondhand smoke exposure (SHSe) alters serum arachidonic acid (AA) metabolism in cystic fibrosis (CF). Serum lipidomics were measured by high-performance liquid chromatography-tandem mass spectrometry in a combined group of infants and children with CF. (A) SHSe was associated with a significant decrease in serum AA metabolites, specifically: prostaglandin D2 (PGD2), leukotriene C4, 8S-hydroxy-5Z, 9E, 11Z, 14Z-eicosatetraenoic acid (8(S)-HETE), 9S-hydroxy-5Z,7E,11Z,14Z-eicosatetraenoic acid (9(S)-HETE), 12(S)-hydroperoxy tetraenoic eicosatetraenoic acid (12(S)-HETE) and 14(15)-epoxy-5Z,8Z,11Z-eicosatrienoic acid (14(15)-EET). (B) Decreased PGD2 concentrations were associated with increased hospitalisations (p=0.007, Wilcoxon rank-sum test) and (C) decreased weight z score (r=0.34, p=0.008). (D) PGD2 was not associated with FEV1 (r=0.19). Associations between other AA metabolite concentrations and hospitalisations, weight z score and FEV1 were not significant. PGD2 and other AA metabolite concentrations also did not correlate with height z score or oropharyngeal/sputum bacterial cultures (not shown). Correlations were performed using Pearson’s correlation coefficient.
Altered metabolite concentrations were then correlated with clinical outcomes in all 57 infants and children with CF. Decreased PGD2 concentrations were significantly associated with a history of hospitalisation for pulmonary exacerbation in the past 6 months (p=0.007, figure 2B) and correlated with lower weight z scores (r=0.34, p=0.008, figure 2C), but not FEV1 (r=0.19, p=0.14, figure 2D). Other associations between AA metabolite concentrations and clinical outcomes, including height z score and oropharyngeal/sputum bacterial cultures, were not significant.
The original cohort had few infants with CF without SHSe (n=4), thus we enrolled a second cohort of 20 infants with CF at two institutions (NCH and RCH) with 40% SHSe determined via hair nicotine concentration. We then measured serum PGD2 concentrations in this second cohort. This validation strategy confirmed that infants with CF exposed to SHSe had significantly decreased serum PGD2 concentrations compared with infants not exposed to SHSe, in a similar pattern compared with the original cohort (online supplement table E1).
SHSe alters gene expression profiles in CF
To further examine the impact of SHSe on inflammatory/immune and prostaglandin gene signalling, we analysed whole blood transcriptional profiles in 28 children 1–10 years of age with CF (10 without SHSe, 18 with SHSe) and 12 age-matched healthy controls (demographics listed in the online supplement table E2). The limited number of infants with CF (n=4) with blood available for transcriptional profile analysis and no SHSe precluded further analyses in this age group. Class comparisons using linear mixed models adjusted for age and race (p<0.01; Benjamini-Hochberg corrected and ≥1.25-fold change) identified 2137 differentially expressed transcripts between children with CF exposed to SHSe and age-matched healthy controls (figure 3A). On the other hand, there were only 224 differentially expressed transcripts between children with CF and no SHSe compared to controls . These transcripts were combined using a Venn diagram that yielded a total of 2188 transcripts identified in children with CF with and without SHSe compared with healthy age-matched controls (online supplement figure E1). Of those, 50% were overexpressed and 50% underexpressed. The top 10 overexpressed and underexpressed transcripts in children with CF according to SHSe are listed in the online supplement tables E3 (non-SHSe) and E4 (SHSe), respectively. In children with CF without SHSe, haptoglobin (inhibits oxidative activity of free haemoglobin) and RFX2 (regulatory gene that binds DNA) were the top overexpressed genes, while Fc receptor like 6 (acts as a major histocompatibility complex class II receptor) and CD8 antigen (mediates efficient cell-cell interactions between cytotoxic T lymphocytes and other immune components) were the top underexpressed genes.

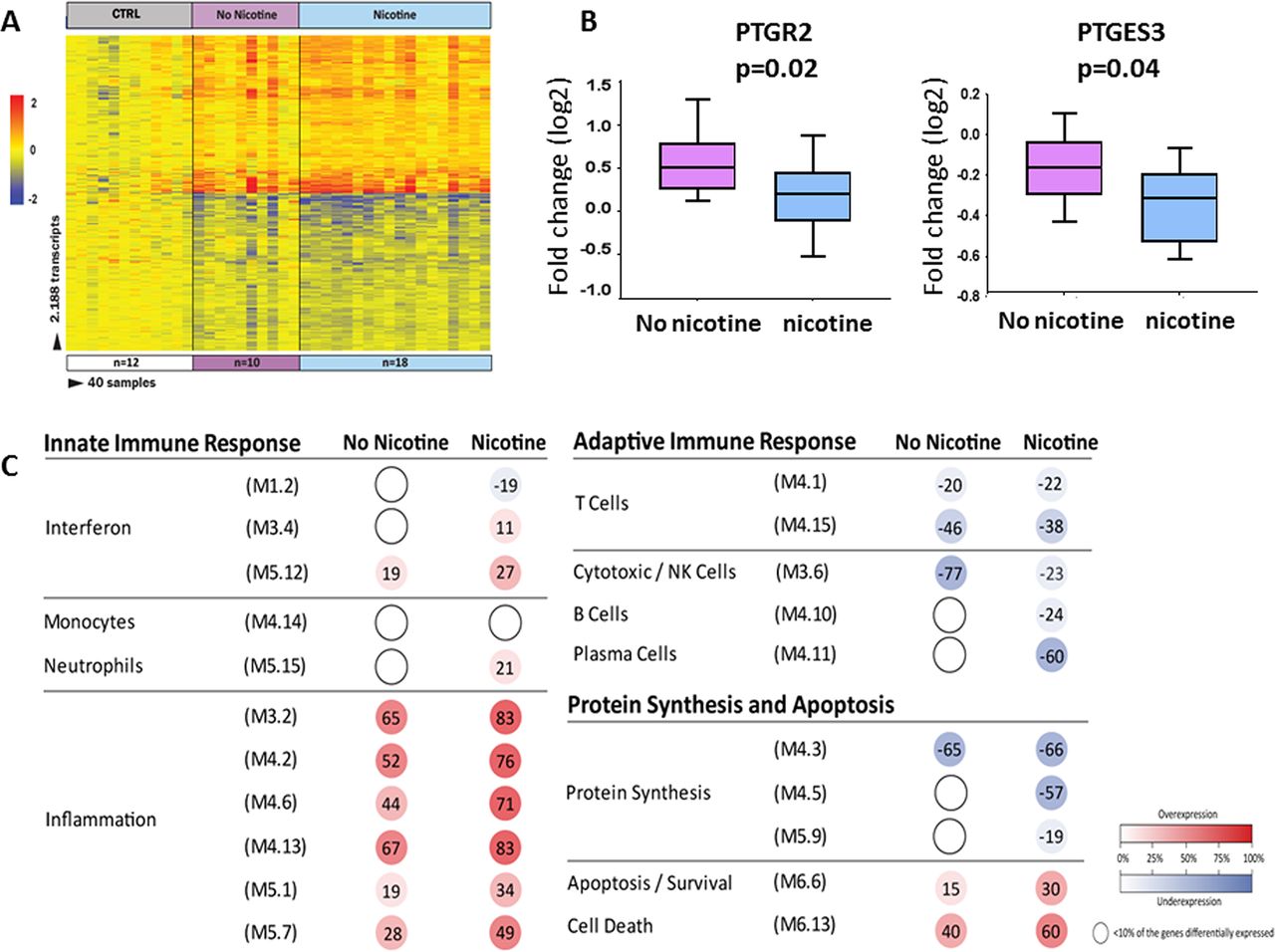
Secondhand smoke exposure (SHSe) alters whole blood transcriptional profiles in cystic fibrosis (CF). (A) Class comparisons using linear models adjusted for age and race (p<0.01; Benjamini-Hochberg corrected flase discovery rate (FDR) and ≥1.25-fold change), in children with CF and SHSe (n=18) vs age-matched healthy controls (n=12) and children with CF and non-SHSe (n=10) vs the same healthy controls, identified 2188 differentially expressed transcripts that were present in either of these conditions. Infants were not analysed. Transcripts are organised in a heatmap format where each row represents a single transcript and each column represents a subject sample. Red colour indicates overexpression and blue colour underexpression of a transcript compared with the median expression of healthy controls (yellow). (B) Two prostaglandin transcripts (PTGR2 and PTGES3) were found to be significantly underexpressed in children with CF exposed to SHSe as compared with those not exposed to SHSe. (C) Modular expression in children with CF exposed to SHSe (n=18) and not exposed to SHSe (n=10), each group was compared with matched healthy controls (n=12). Children with CF exposed to SHSe demonstrated significant overexpression of inflammation genes and greater suppression of protein synthesis and plasma cell-related transcripts. The intensity of the modules (dots) indicates the proportion of overexpressed (in red) or underexpressed (in blue) transcripts within each module. Numeric values indicate the exact percentage of transcripts expressed in each specific module. A blank dot indicates that <10% of the genes in the module were differentially expressed.
In contrast, in children with CF and SHSe, matrix metallopeptidase 9 (MMP-9, involved in the breakdown of extracellular matrix) and regulatory factor X, 2 (RFX2, transcriptional activator) were the top overexpressed genes, while eukaryotic translation initiation factor 3 subunit C like (EIF3CL, binds the 40S ribosome and mRNAs to enable translation initiation) and vanin 2 (VNN2, involved in transendothelial migration of neutrophils) were the top underexpressed genes. In addition, we identified two AA-derived prostaglandins genes, prostaglandin reductase 2 (PTGR2) and prostaglandin E synthase 3 (PTGES3) that were significantly greater underexpressed in children with CF exposed to SHSe compared with children with CF not exposed to SHSe (figure 3B). These prostaglandin genes have been related to activation of the peroxisome proliferator-activated receptor (PPAR) and prostaglandin E synthesis, respectively.
In order to define the biological significance of the SHSe profile, modular analyses were conducted separately in children with CF with and without SHSe in relation to non-CF, age-matched, healthy controls (figure 3C, online supplement table E5). Transcriptional modules represent groups of differentially expressed genes sharing a similar function as described.27 28 In children with CF, the presence of SHSe was associated with marked overexpression of inflammation-related genes, significant underexpression of one of the interferon modules (M 1.2) and with greater suppression of protein synthesis and plasma cell-related genes compared with children with CF without SHSe. Both CF groups showed underexpression of NK cell-related genes compared with controls, although to a lesser extent in the SHSe. Overall, these findings suggest SHSe differentially influences immune and inflammatory responses in children with CF, specifically with enhanced expression of inflammation-related genes and suppression of specific prostaglandin-related genes.
In vitro production of arachidonic acid metabolites is altered in CF monocyte-derived macrophages after SHSe
PGD2 activates non-CF macrophage effector functions.29 To corroborate serum PGD2 findings and interrogate PGD2 production in primary CF macrophages, we used an in vitro model of SHSe involving exposure to CSE. These experiments were conducted using macrophages from children and adults with and without CF due to the limited available blood volume in infants. First, macrophage cell death was measured in response to CSE to verify optimal exposure based on non-CF literature. Concentrations of >15% CSE were associated with increased macrophage cell death as measured by lactate dehydrogenase (LDH) release (figure 4A). We then measured CF macrophage PGD2 concentrations after exposure to varying concentrations of CSE. CF MDMs demonstrated a dose-dependent decrease in PGD2 concentrations with increasing CSE concentrations (figure 4B). Western blot analysis of MDM lysates confirmed decreased expression of PGD2 in CF MDMs in response to SHSe (figure 4C). Zero (control) and 15% CSE was chosen for further SHSe studies based on observed cellular toxicity and cell tolerance.

Secondhand smoke exposure (SHSe) alters cystic fibrosis (CF) macrophage prostaglandin D2(PGD2) production. (A) Cytotoxicity as %LDH release was measured in child and adult CF monocyte-derived macrophage supernatants in response to increasing concentrations of cigarette smoke extract (CSE), 24 hours challenge, n=3. (B) Changes in the arachidonic acid metabolite PGD2 were measured in CF macrophage supernatants in response to increasing concentrations of CSE during a 24 hours challenge. (C) PGD2 expression was determined by western blot analysis in CF and non-CF macrophages in response to exposure to a 0% or 15% CSE, n=3, representative image shown.
Next, we attempted to validate overall serum AA profile responses to SHSe by measuring macrophage production of total AA metabolites. Lipidomics was performed using supernatants from CF and non-CF macrophage exposed to either 0% or 15% CSE. Except for 8(S)-HETE that was comparable between CF and non-CF MDM, SHSe was associated with significantly less production of PGD2, 9(S)-HETE, 15(S)-HETE, lipoxin A4, 12(S)-HETE, leukotriene E4, and prostaglandin E2 (PGE2) in CF compared with non-CF MDMs (figure 5). Overall, these results confirmed the results from serum samples (figure 2) and indicated a lack of or reduced production of AA metabolites in response to SHSe only in CF MDMs.

Secondhand smoke exposure alters arachidonic acid (AA) metabolism in cystic fibrosis (CF) macrophages. Lipidomics for AA metabolites was performed via high-performance liquid chromatography-tandem mass spectrometry in CF and non-CF macrophage supernatants from children and adults after exposure to either 0% or 15% cigarette smoke extract (CSE), n=3, 24 hours challenge. *p<0.05, t-test. LTE4, leukotriene E4; LXA4, lipoxin A4; PGE2, prostaglandin E2; PGD2, prostaglandin D2; 8(S)-HETE, 8S-hydroxy-5Z,9E,11Z,14Z-eicosatetraenoic acid; (9(S)-HETE, 9S-hydroxy-5Z,7E,11Z,14Z-eicosatetraenoic acid; 12(S)-HETE, 12(S)-hydroperoxy tetraenoic eicosatetraenoic acid.
Last, to determine if SHSe specifically impacted macrophage bacterial clearance, we analysed the effect of SHSe on in vitro bacterial clearance/killing in CF and non-CF MDMs. MDMs from all groups were infected in vitro with bacterial clinical isolates from patients with CF including MRSA, P. aeruginosa, and Burkholderia cenocepacia, and bacterial loads measured according to CSE and CF status. In non-CF MDMs P. aeruginosa, B. cenocepacia and MRSA bacterial loads were comparable independent of exposure to CSE (figure 6A-C). In CF MDMs, significant increases in MRSA, but not P. aeruginosa or B. cenocepacia, bacterial loads were identified in response to CSE (figure 6A). Therefore, combining the serum, whole blood and in vitro data, we found a direct effect of SHSe on AA metabolite production, and in particular PGD2 production, at the systemic and immune cell (macrophage) level in patients with CF that was associated with impaired MRSA clearance.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Secondhand smoke exposure decreases methicillin-resistant Staphylococcus aureus (MRSA) killing in cystic fibrosis (CF) macrophages. Human CF and non-CF macrophages from children and adults were subacutely (72 hours) treated with 15% cigarette smoke extract (CSE) and infected with a clinical isolate of either (A) MRSA, (B) Pseudomonas aeruginosa or (C) Burkholderia cenocepacia, n=6. Bacterial counts were measured via colony-forming unit (CFU) assay and presented as CFU/mL of media.
Discussion
CF lung disease begins in early life with inflammation, infection and structural and functional changes that often precede respiratory symptoms. Thus, therapeutic interventions should be focused on preventive measures, rather than improving established disease. Nevertheless, CF is a heterogeneous disorder and deficits exist in predicting both the onset and progression of early lung disease despite well-characterised genotyping. These deficits are crucial since early treatment is essential to prevent permanent pulmonary damage. Although inflammation plays a prominent role in CF pathogenesis, the contribution of environmental factors including SHSe to the infectious and inflammatory cycle remains poorly understood. In this study, we showed that infants and young children with CF have high rates of exposure to SHSe as assessed objectively, with associated altered inflammatory gene expression and dysregulated AA metabolism, together resulting in impaired bacterial clearance. These findings have important clinical implications for the clinical care of young children with CF.
In addition to knowledge deficits in the extent of SHSe in early CF, little is known about the impact of SHSe on early pathologic mechanisms such as inflammation in the respiratory tract or the failed resolution of this enhanced inflammation. Specific biochemical mechanisms of SHSe in CF have not been well characterised. In adults without CF, primary cigarette smoking causes systemic CFTR dysfunction.30 31 In adults without CF, smoking is also associated with alterations in AA metabolism and phospholipid hydrolysis including prostaglandins such as PGE2,8 and smoke exposure inhibits alveolar macrophage production of AA metabolites involved in inflammation.32 In turn, chronic inflammation and immune dysregulation are key factors in CF pathogenesis including early onset release of pro-inflammatory mediators and excess neutrophilic lower airway inflammation independent of infection.33–35 Failed resolution of chronic inflammation in CF has not been studied in the context of SHSe, but is suggested by baseline alterations in AA metabolites and their relation to poor growth.36 Dysregulated AA metabolism has also been demonstrated during CF exacerbations and periods of poor growth.10 11 Changes in fatty acid metabolism (AA metabolism in particular) have been shown in multiple CF cohorts at baseline37 38 and are partially corrected by CFTR modulators.39 Combined, these changes indicate the potential role of CFTR in influencing AA metabolism in CF. SHSe may worsen these baseline changes in CF AA metabolism, particularly in young infants and children. However, SHSe has not been objectively measured in prior studies and its role as a primary driver of earlier reported changes in CF AA metabolism has not been determined conclusively. Furthermore, characterisation of SHSe’s impact on specific fatty acids such as linoleic acid and docosahexaenoic acid that were not measured in this study are important to determine in future CF studies due to their observed alterations independent of SHSe.11 37 40 41
Functional AA metabolism is critical in both the onset and the resolution of inflammatory responses. Nicotine exposure was associated with reduced 15-lipoxygenase (LO)-mediated pathway metabolites (HETEs), which play an integral role in the switch of eicosanoid biosynthesis from leukotrienes to lipoxins. Alterations of this pathway have been previously demonstrated in children with CF and are suggested to play a role in the failed resolution of inflammation.42 However, a relation of the LO pathway to SHSe in CF has not been previously shown. Additionally, PGD2, an upstream activator of PPAR-γ, is significantly decreased by SHSe and associated with worsened clinical outcomes, suggesting further impact on the resolution of inflammation.43 PPAR-γ has been studied in CF as a potential therapeutic target that modulates the immune response in CF,44 thus implicating SHSe as a potential modifier of this reported response. In our study, PGD2 alterations were identified at the systemic (whole blood and serum) and cellular (macrophage) level, suggesting its robustness as a potential biomarker of early disease responses related to SHSe in CF. Decreases in AA metabolite production may also represent general damage to cells which normally produce these metabolites, as evidenced by our human macrophage data.
Early immune responses to bacteria are poorly defined in CF, particularly in the context of SHSe, which has been shown to decrease host phagocytic responses in subjects without CF.45 In our study, SHSe was associated with decreased macrophage-mediated bacterial killing of the common CF pathogen MRSA as well as increased clinical acquisition of P. aeruginosa and S. maltophilia. Our data support recent work that demonstrated early postnatal SHSe in a murine muco-obstructive airway model alters immune function and disrupts bacterial clearance of P. aeruginosa.46 Smoke exposure has been previously shown to augment expression of MRSA virulence factors in non-CF murine models,47 48 and impact phagocytosis of P. aeruginosa in CF49 and non-CF cells in a CFTR-dependent manner.50 Early P. aeruginosa acquisition predicts long-term outcomes in children with CF,51 52 thereby predicting that detection and prevention of SHSe in young children with CF can aid in delaying establishment of certain chronic bacterial infections and subsequently worsened outcomes. The lack of impact of SHSe on macrophage-mediated B. cenocepacia clearance may be related to inherent differences in pathogenicity between B. cenocepacia and other pathogens in CF, such as differential regulation of the oxidative burst.53 Likewise, differences in increased clinical acquisition of P. aeruginosa in children with SHSe compared with macrophage-mediated clearance suggests that other immune cells responsible for P. aeruginosa clearance such as neutrophils may be impacted by SHSe.
In addition to an impact on AA metabolism and AA gene expression, SHSe in children with CF appears to differentially regulate gene expression related to inflammation and both innate and adaptive immunity pathways. A robust overexpression in pathways related to inflammation gene expression and cell death was observed in children with CF exposed to SHSe, essentially exaggerating the profile of heightened inflammation present in children with CF compared with those without CF. In particular, MMP-9 was the top over-expressed gene identified in children with CF exposed to SHSe. PGD2’s downstream metabolites have been associated with suppression of MMP-9 expression,54 further strengthening the proposed connection between PGD2 and altered inflammation during SHSe in patients with CF. MMP-9 is also associated with bronchiectasis progression in early CF lung disease,55 identifying SHSe as a potential modifier of structural lung damage.
Although dysregulated AA metabolism is involved in many pathways of inflammation, other pathways are likely involved during SHSe as noted in the gene modular analysis. Genes related to plasma cell function were underexpressed, suggesting an additive suppression of SHSe on adaptive immune responses. Connections between AA signalling and plasma cell function outside of CF are still being defined, but studies suggest that the activation or downregulation of B cells depends on the specific AA metabolites involved.56 57 Dietary AA intake may also play a key role in B cell activation in infants.58 Several genes related to interferon signalling were also altered with SHSe. Combined, findings from the modular analysis along with the current literature suggest that SHSe early in life critically regulates immune function and may further enhance the predisposition of children with CF to bacterial infections.
Importantly, our cohort had a higher incidence of SHSe compared with data available in the CF Foundation (CFF) Patient Registry.59 However, CFF Patient Registry reporting is based on parental report and not on objective measurements, and therefore may be reflective of under-reporting or a social desirability bias. Additionally, we used hair nicotine to detect SHSe, which allows for a more accurate reflection of long-term exposure. Our data are more closely aligned with recent data from the Early Pseudomonas Infection Control (EPIC) observational study, which reported subjective SHSe in 52% of children over the 3 months prior to study inclusion.50 Regardless of reporting measure, our data suggest that young infants and children with CF are frequently exposed to tobacco products and greatly impacted by such exposures.
This study has limitations. Clinical samples were collected at a single time point, and thus we could not assess for changes in biochemical measures over time. As previously mentioned, we did use a long-term measure of SHSe (hair nicotine), which eliminates potential bias from brief or short-term exposures to tobacco products. The original cohort was limited to a single collection site, and therefore results may not be generalisable to other populations. We did however, confirm AA metabolite findings and detection of hair nicotine in an additional cohort of infants that included a second collection site. Furthermore, the paucity of data regarding SHSe responses in infants and young children with CF lends relevance to this study. Our assessment of bacterial culture positivity was limited to oropharyngeal cultures in this young population who did not routinely produce sputum or undergo bronchoalveolar lavage. Follow-up studies on the role of SHSe in the developing CF microbiome are underway.
In summary, infants and young children with CF demonstrate high levels of quantifiable SHSe associated with both altered inflammatory gene expression and dysregulated AA metabolism resulting in impaired bacterial clearance. Our findings warrant further investigation into how to prevent and halt early disease progression attributable to SHSe in the young population with CF.
Acknowledgments
The authors would like to thank Melinda Smith and Misty Thompson and all of the Clinical Research Services staff for assistance in sample procurement, Sara Mertz for sample processing. The authors would also like to thank all patients and their families for participation.
References
Footnotes
Patient consent for publication Not required.
Contributors BTK, RT, JAG and AM designed the study. BTK wrote the manuscript. BK, AD, DT and BS performed statistical analysis. BTK, BS, RT, DBS, DH, FWW, OR, JAG and AM analysed and interpreted data. JK, RK, CS, AD, BS and LR created experimental data. BTK, RT and DBS collected clinical samples. All authors contributed to editing and approved the final manuscript.
Funding This work was supported in part by The Ohio State University College of Medicine Roessler research scholarship (JK), a Nationwide Children’s Hospital intramural grant (BTK, RT, AM), an American Academy of Pediatrics Julius B. Richmond Center New Investigator Grant (BTK) and CTSA grant UL1TR001070.
Competing interests None declared.
Ethics approval This study was approved by the Institutional Review Boards at Nationwide Children’s Hospital (IRB #12–00084) and Riley Children’s Hospital (IRB #1607531974).
Provenance and peer review Not commissioned; externally peer reviewed.