Article Text

Abstract
Background Pseudomonas aeruginosa lung infections are a huge problem in ventilator-associated pneumonia, cystic fibrosis (CF) and in chronic obstructive pulmonary disease (COPD) exacerbations. This bacterium secretes virulence factors that may subvert host innate immunity.
Objective We evaluated the effect of P. aeruginosa elastase LasB, an important virulence factor secreted by the type II secretion system, on ion transport, innate immune responses and epithelial repair, both in vitro and in vivo.
Methods Wild-type (WT) or cystic fibrosis transmembrane conductance regulator (CFTR)-mutated epithelial cells (cell lines and primary cells from patients) were treated with WT or ΔLasB pseudomonas aeruginosa O1 (PAO1) secretomes. The effect of LasB and PAO1 infection was also assessed in vivo in murine models.
Results We showed that LasB was the most abundant protein in WT PAO1 secretomes and that it decreased epithelial CFTR expression and activity. In airway epithelial cell lines and primary bronchial epithelial cells, LasB degraded the immune mediators interleukin (IL)-6 and trappin-2, an important epithelial-derived antimicrobial molecule. We further showed that an IL-6/STAT3 signalling pathway was downregulated by LasB, resulting in inhibition of epithelial cell repair. In mice, intranasally instillated LasB induced significant weight loss, inflammation, injury and death. By contrast, we showed that overexpression of IL-6 and trappin-2 protected mice against WT-PAO1-induced death, by upregulating IL-17/IL-22 antimicrobial and repair pathways.
Conclusions Our data demonstrate that PAO1 LasB is a major P. aeruginosa secreted factor that modulates ion transport, immune response and tissue repair. Targeting this virulence factor or upregulating protective factors such as IL-6 or antimicrobial molecules such as trappin-2 could be beneficial in P. aeruginosa-infected individuals.
- airway epithelium
- bacterial infection
- cystic fibrosis
- innate immunity
- respiratory infection
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is the key question?
To study mechanistically the deleterious effect of Pseudomonas aeruginosa LasB protease, an important bacterial virulence factor present in infected patients with cystic fibrosis (CF) and patients with chronic obstructive pulmonary disease (COPD) in an infectious and inflammatory setting.
What is the bottom line?
This is the first demonstration that overexpression of interleukin (IL)-6 in the lungs promotes resistance against P. aeruginosa infection, through the upregulation of an IL-17/IL-22 antimicrobial and repair pathway.
Why read on?
By showing that IL-6 and trappin-2 can rescue the host from P. aeruginosa LasB deleterious activity, our novel findings hold important implications for both genetically CFTR-sufficient individuals infected with P. aeruginosa (eg, during nosocomial infections, or during COPD exacerbations), as well as for individuals with CF.
Introduction
Cystic fibrosis (CF) is the most common genetically inherited disease in Caucasian populations (1 in 3500 newborns) and 70%–90% of CF individuals harbour the F508del mutation, resulting in misfolding and incorrect trafficking of the cystic fibrosis transmembrane conductance regulator (CFTR) molecule to the epithelial membrane.1–3 CFTR is an anion channel that regulates fluid homeostasis on epithelial surfaces.4 In the airways, a loss of function and/or stability3 of this protein is thought to sequentially induce hypohydration, mucus accumulation, bacterial infections (eg Pseudomonas aeruginosa, Burkholderia cepacia) and chronic inflammation via the recruitment of neutrophils.5 Although CFTR mutations are responsible for disease in individuals with CF and lead to chronic P. aeruginosa infections (a key hallmark of the disease), CFTR expression has recently also been reported as downregulated in epithelial cells treated with cigarette smoke.6 7 In vivo also, genetically CFTR-sufficient cigarette smokers showed a decrease in CFTR function,6 7 and similarly, in biopsies from non-CF chronic obstructive pulmonary diseases (COPD)/emphysemateous patients, CFTR protein expression was significantly decreased with disease activity and was associated with inflammation.8 We have shown recently that neutrophil elastase (NE) is able to degrade CFTR in vitro and in vivo, through the activation of intracellular calpains, potentially explaining infectious and inflammatory exacerbations in CF and COPD.9 Importantly, when comparing the effect of Pseudomonas infection on CFTR in wild-type (WT) and NE−/− mice, we found NE to account for only part of CFTR degradation. We therefore hypothesised that other factors, of P. aeruginosa origin, may also target and have deleterious effects on CFTR and on innate immune responses downstream of CFTR. Here, we identify LasB, a P. aeruginosa type II secretion system metalloprotease and an important virulence factor present in CF secretions10–13 as the main secreted protein in the secretome from the pseudomonas aeruginosa O1 (PAO1) strain. Using biochemical and functional assays, we investigated its effects on CFTR function and on the regulation of innate immune responses, in vitro and in vivo.
We demonstrate that LasB degrades CFTR and downregulates an interleukin (IL)-6–antimicrobial–lung repair pathway in vitro and ex vivo in primary airway cells from patients. Furthermore, we show that this pathway can be rescued in vivo in mice by overexpressing IL-6 and the antimicrobial molecule trappin-2.
Because P. aeruginosa infections are a common feature in CF and COPD/emphysema exacerbations,14 and since LasB is invariably found in inflammatory secretions (particularly in CF10–13), our data suggest that targeting P. aeruginosa LasB and/or stimulating the IL-6/antimicrobial/repair axis maybe an interesting and novel approach for tackling the inflammatory processes underlying P. aeruginosa-induced lung disease exacerbations.
Materials and methods
Materials
Phosphoramidon (PA, R7385), amiloride, forskolin, IBMX and CFTRInh172 were obtained from Sigma-Aldrich. Purified LasB (pLasB) was a kind gift from Pr. Gerd Döring and recombinant human IL-6 and 1β were purchased from R&D Systems. Tace II substrate was obtained from Enzolife Science.
Preparation and analysis of WT PAO1 and ΔLasB PAO1 secretomes (WT-SEC and ΔLasB -SEC)
PAO1 WT, a strain expressing LasB,15 and PAO1 ΔlasB strain (gift from Pr. D. Ohman) were grown overnight in Luria Broth (LB) medium (1% Bactotryptone, 0.5% Bacto Yeast Extract, 0.5% NaCl) under agitation. Bacterial suspensions were then centrifuged at 4000 g (15 min, 4°C), 6000 g (10 min, 4°C) and 12 000 g (10 min, 4°C) before the remaining supernatants (secretomes (SEC)) were filter-sterilised using 0.2 µm pore syringe filters, aliquoted and stored at −80°C until use.
Purified SECs were then analysed by sodium dodecyl sulfate (SDS-PAGE) or zymography. For the latter, SECs were prepared in β-mercapto-ethanol-free 4× Laemmli buffer and separated on 7% acrylamide gels containing 2% gelatin. After migration, gels were washed and incubated overnight in the developing buffer, stained in 0.05% Coomassie brilliant blue for 1 hour and destained with appropriate Coomassie R-250 destaining solution (methanol:acetic acid:water (50:10:40)).
Cytokine and trappin-2 secretion measurements
Media were collected and assayed for IL-8 and IL-6 by ELISA kits (R&D Systems, Minneapolis, Minnesota, USA) following the manufacturer’s instructions. Human trappin-2 was measured using an ELISA available in-house (Ref S4).
Cells, cell culture and analysis, scratching protocols, short-circuit current measurements, adenovirus constructs
These are described in detail in the online supplementary file 1.
Supplementary file 1
In vivo experiments
Procedures involving mice were approved by our local ethical committee (Paris-Nord/No 121) and by the French Ministry of Education and Research (agreement number 04537.03). Eight-week-old male C57BL/6 mice and trappin-2 transgenic mice (hereafter called eTg mice) were from Janvier (Le Genest-Saint-Isle, France) and generated by our group,16 respectively. Mice were anaesthetised using intramuscular injection of ketamine 500 and xylazine 2% in 0.9% NaCl (20:10:70). Different quantities of pLasB, Ad-vectors or PAO1 bacteria were instilled either intranasally or intratracheally (final volume of 40 µL max), followed by either monitoring their survival, or by humanely killing the animals (overdose of 100 µL intraperitoneally injected pentobarbital) for mechanistic studies (see online supplementary file 1). Bronchoalveolar lavages (BALs) fluid were obtained by cannulating the trachea and instilling 2×1 mL of phosphate-buffered saline (PBS). Typically, a volume of 1.7 mL of BALF was retrieved and centrifuged at 2000 rpm for 10 min. Supernatants were used for protein, cytokine (ELISA) and haemoglobin (used as a surrogate for lung damage, absorbance reading at 405 nm) measurements. Cell pellets were used for cell differential analysis (Diff-Quick, Dade Diagnostika GmbH, Unterschleissheim, Germany).
In parallel, RNA isolation, followed by reverse transcription and quantitative PCR (RT-q-PCR), was performed as described in online supplementary file 1. Finally, lung tissue was also used for quantifying bacteria after plating extracts on agarose plates.
Statistical analysis
Data were analysed with GraphPad Software with either one-way analysis of variance (followed by Bonferroni post hoc tests) or non-parametric analysis, followed by post-Dunn’s test for multiple comparisons.
Survival curves in murine models experiments were plotted using Kaplan-Meier curves and statistical tests were performed using the Log-rank (Mantel-Cox) test.
Results
LasB is the main protein in the P. aeruginosa secretome, degrades epithelial cells CFTR and decreases CFTR activity in vitro
We first demonstrated, using Coomassie blue staining of SDS-PAGE gels and zymography techniques, that LasB was the major protein secreted in WT PAO1 secretome and that it was active as a metalloprotease (see relevant information on in online supplementary file 2).
Supplementary file 2
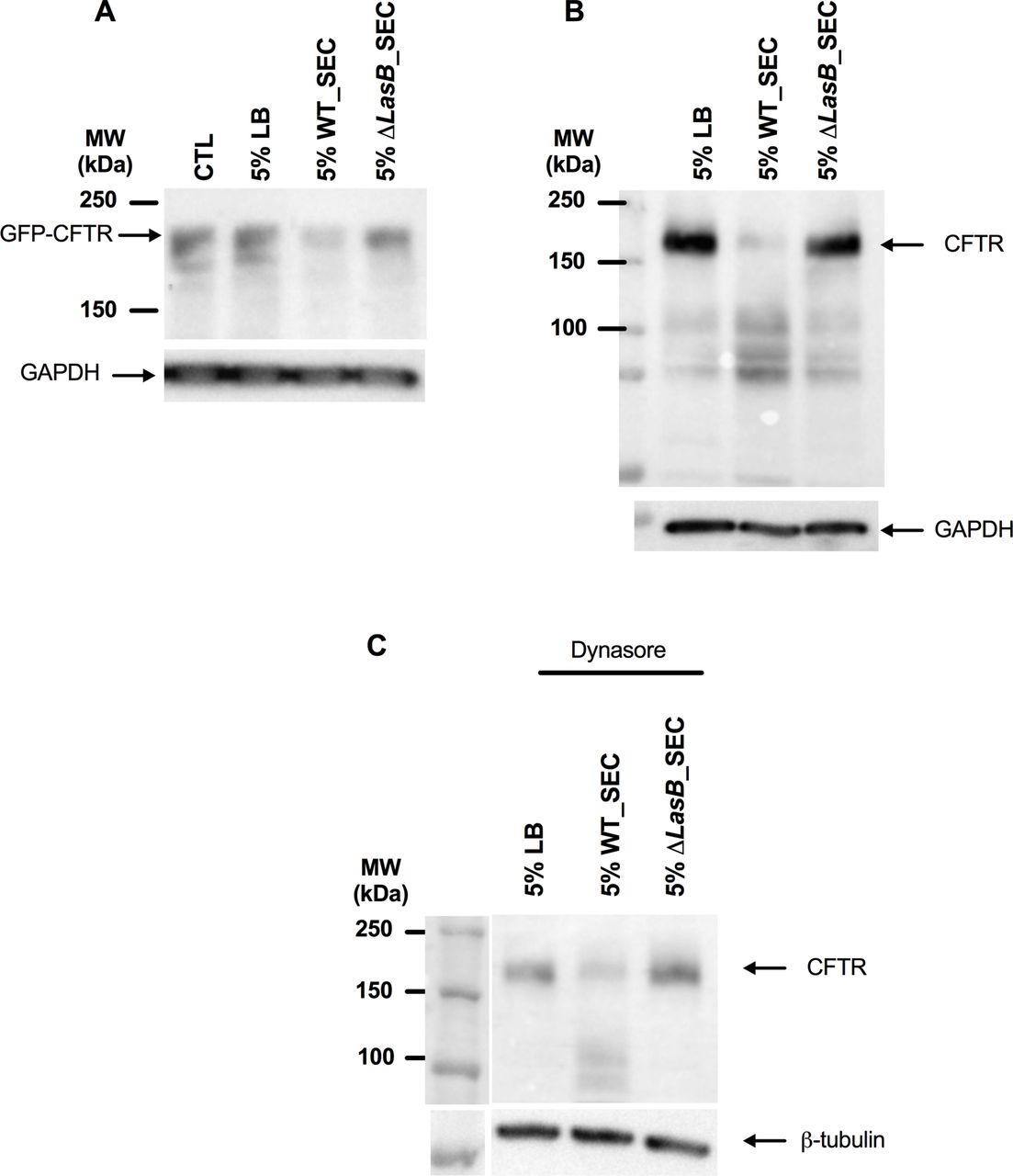
We then investigated the effect of PAO1 WT and ΔLasB-SEC on CFTR expression in different cell lines. In NCI-H292 cells (cells expressing minimal amounts of endogenous CFTR) transfected with Ad-GFP-WT-CFTR, PAO1 WT SEC (but not ΔLasB-SEC) induced CFTR degradation (figure 1A). Similar results were observed in polarised Calu-3 cells, which express high amounts of endogenous CFTR (figure 1B). This was not due to an increase in CFTR endocytosis as these results were reproduced in the presence of dynasore (figure 1C).

LasB downregulates CFTR protein levels in epithelial cells. (A) Western blot analysis of CFTR expression in NCI-H292 cells overexpressing GFP-WT-CFTR, as detected by anti-GFP antibody after a 24-hour treatment with either 5% LB medium, 5% WT-SEC or 5% ΔLasB-SEC. (B) Western blot analysis of CFTR expression in polarised Calu-3 cells after a 24-hour treatment with either 5% LB medium, 5% WT-SEC or 5% ΔLasB-SEC, as detected by anti-CFTR(596) antibody. (C) Western blot analysis was performed as in (B), except that cells were preincubated with dynasore (80 µM), a GTPase inhibitor that targets dynamin and blocks endocytosis, at 37°C for 30 min. Each image is representative of n=3 independent experiments. CFTR, cystic fibrosis transmembrane conductance regulator; GAPDH, glyceraldehyde-3 phosphate dehydrogenase; LB, Luria Broth; SEC, secretomes.
We then evaluated if this decrease in expression induced a decrease in activity in WT-CFTR cystic fibrosis bronchial epithelial (CFBE) cells (stably expressing CFTR). WT-SEC (but not ΔLasB-SEC) significantly decreased CFTR activity as shown by the decrease in forskolin/IBMX-induced current (figure 2A, figure 2C positive delta) and CFTRInh172-sensitive current (figure 2D, negative delta). This effect was specific to CFTR as shown by the absence of effect of the SECs on ENaC current (amiloride sensitive current, figure 2A, figure 2B negative delta).
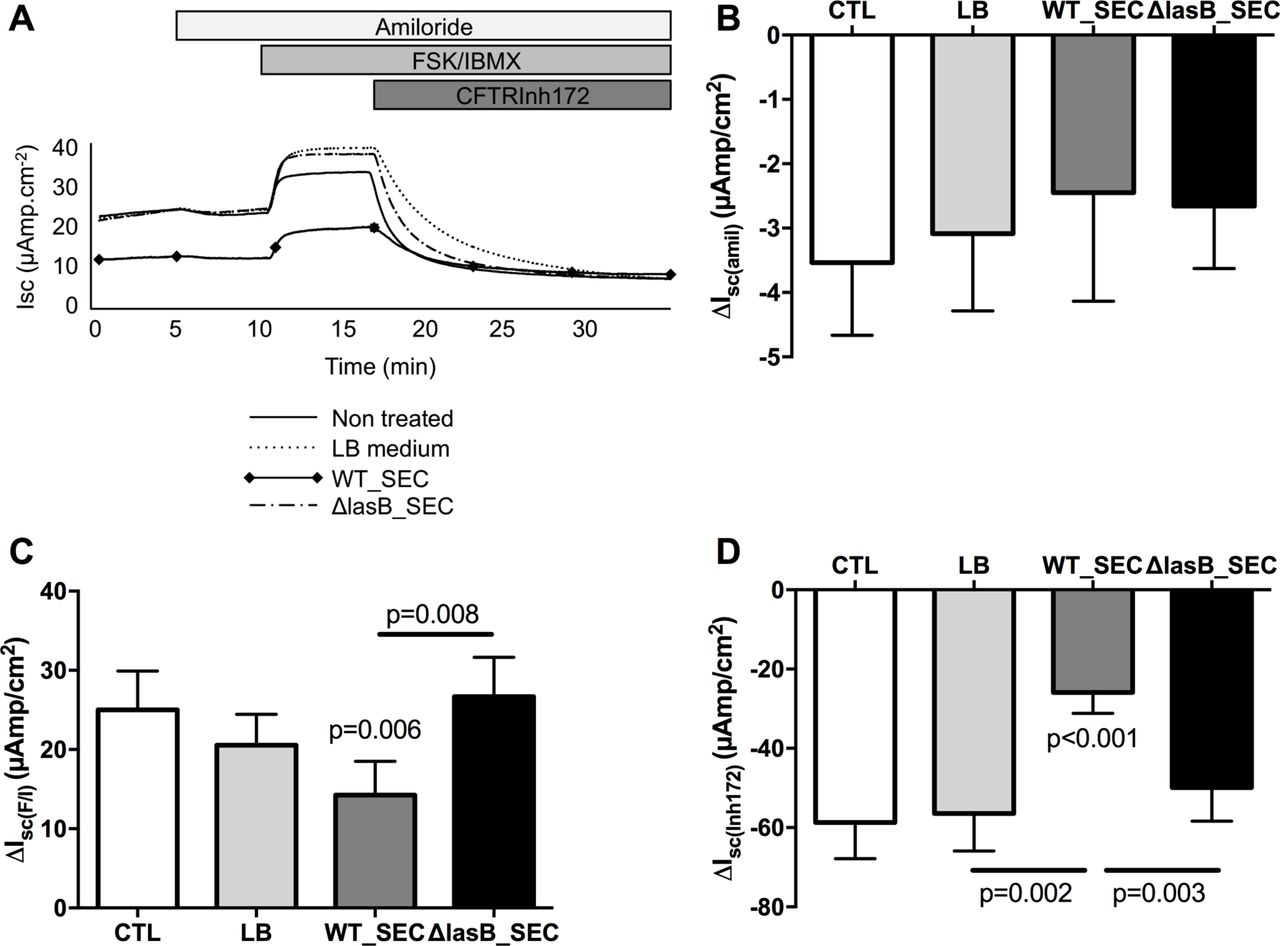
LasB downregulates CFTR activity in WT-CFTR overexpressing CFBE cells. CFBE cells were apically treated for 24 hours with LB, WT-SEC or ΔLasB-SEC dilutions in MEM. When mounted in Ussing chambers, currents were allowed to stabilise and the changes in short-circuit currents (ΔIsc) were measured in response to amiloride (100 µM, apical), a forskolin (FSK, 10 µM)/isobutylmethylxanthine (IBMX, 100 µM) cocktail (apical and basolateral) and CFTRInh172 (5 µM, apical). Panel A shows representative short-circuit recordings of CFBE cells responses after treatment. Panels B and C depict amiloride-sensitive (negative delta) and FSK/IBMX-induced currents (positive delta), respectively. Panel D shows data with CFTRinh172, a specific inhibitor of CFTR (negative delta). Results are shown as mean±SEM Statistics: (C): one-way analysis of variance (ANOVA) and Dunn’s post-test, n=8; (D): one-way ANOVA and Dunn’s post-test, n=12. CFBE, cystic fibrosis bronchial epithelial; CFTR, cystic fibrosis transmembrane conductance regulator; LB, Luria Broth; SEC, secretomes.
LasB downregulates IL-6 and the antimicrobial molecule trappin-2 in human lung epithelial cells
It has previously been suggested that the absence of CFTR ‘per se’ may provide an ‘inflammatory phenotype’, with modulated secretion of IL-6 and IL-8 for example17 as well as that of antimicrobial molecules.18–21 In addition, downregulation of CFTR affects acid/base homeostasis of epithelial surfaces and can therefore also affect antimicrobial function.22 Our results shown above therefore prompted us to test whether LasB might modulate these important epithelial cell innate immune mediators.
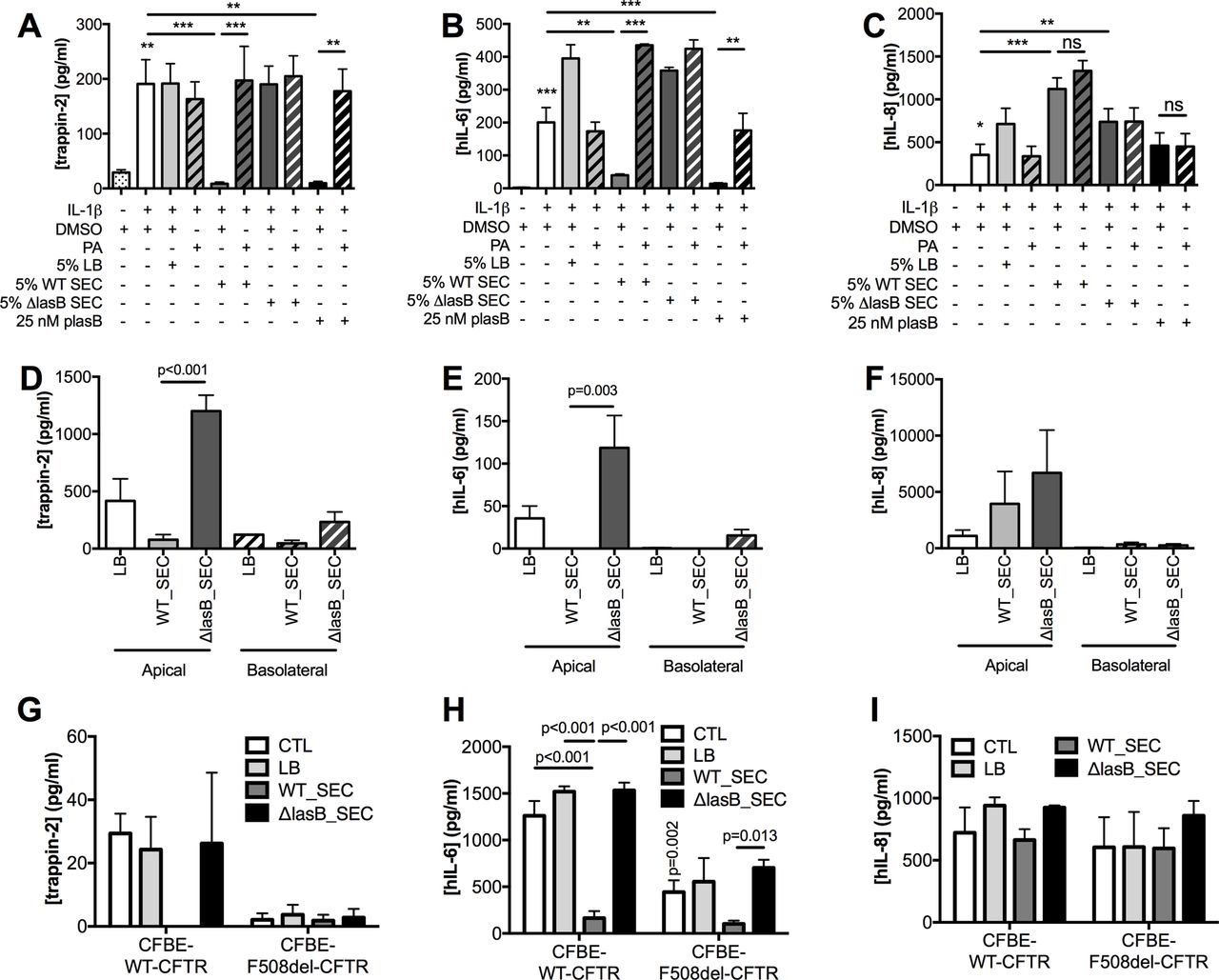
Because NCI-H292 produced low amounts of trappin-2 at steady state, its production was upregulated with IL-1β (figure 3A–C), whereas no upregulation was needed in Calu-3 (figure 3D–F) and CFBE cells (figure 3G–I). In NCI-H292, LB medium + IL-1β and ΔLasB-SEC + IL-1β treatments induced an increase in IL-6 (figure 3B) and IL-8 (figure 3C) compared with IL-1β alone, indicating that LB and secretome components were stimulatory. However, WT-SEC, despite containing the same components as ΔLasB-SEC, completely abolished IL-1β-induced trappin-2 (figure 3A) and IL-6 (figure 3B) secretions but had no effect on IL-8 production (figure 3C). In addition, we showed in these cells that purified pLasB had a similar effect than the WT-SEC and that PA, a metalloprotease inhibitor, inhibited pLasB-induced and WT-SEC-induced downregulation of trappin-2 and IL-6 (figure 3A-B). In Calu-3 cells, ΔLasB-SEC similarly increased trappin-2, IL-6 and IL-8 production (figure 3D-F), compared with LB medium-treated cells, demonstrating as above that the bacterial pathogen associated molecular patterns (PAMPs) contained in ΔLasB-SEC can up-regulate these mediators. By contrast, WT-SEC, again, completely inhibited trappin-2 and IL-6 protein recovery in the supernatants (figure 3D-E), whereas IL-8 protein levels were unaffected (figure 3F).

LasB reduces trappin-2 and IL-6 recovery, but not that of IL-8, in the supernatants of NCI-H292, Calu-3 and CFBE cells (WT-CFTR and ΔF508). (A–C): NCI-H292 cells were preincubated with IL-1β for 1 hour prior to the addition of diluted SEC (5%) or purified LasB (pLasB) for 4 hours, supplemented or not with the metalloprotease inhibitor phosphoramidon (PA, 8.5 µM). DMSO was also added as a control, since it was used as a diluent for PA. Trappin-2, IL-6 and IL-8 protein levels were measured by ELISA in cell supernatants (n=4, *p<0.05, **p<0.01, ***p<0.001, ns: not significant; ANOVA). (D–F): Calu-3 cells were incubated with LB medium or with diluted SECs (5%) for 4 hours. Trappin-2, IL-6 and IL-8 protein levels were measured as above (n=3, ANOVA). (G–I): CFBE-WT-CFTR and CFBE-F508del-CFTR cells were either untreated (CTL), treated with LB medium, or with diluted SEC (1%) for 24 hours and ELISAs performed as above (n=3–4, two-way ANOVA). Results are shown as mean±SEM. ANOVA, analysis of variance; CFBE, cystic fibrosis bronchial epithelial; CFTR, cystic fibrosis transmembrane conductance regulator; DMSO, dimethyl sulfoxyde; IL; interleukin; LB, Luria Broth; SEC, secretomes.
Similar results were obtained from CFBE cells overexpressing WT-CFTR (figure 3G-I) where WT-SEC, but not ΔLasB-SEC, downregulated apical secretion of IL-6 (figure 3H) and trappin-2 (figure 3G), whereas IL-8 levels remained unchanged (figure 3I). Since we have shown above that LasB decreased CFTR expression and activity, it appeared interesting to evaluate the effect of this metalloprotease on the immune mediators in a CFTR-deficient context. Interestingly, when mutant CFTR (F508del-CFTR) overexpressing CFBE cells were compared with WT-CFTR CFBE cells, the latter had higher levels of IL-6 and trappin-2 protein (but not IL-8), compared with the former (figure 3G–I). Notably, as in WT-CFTR cells, WT-SEC (but not ΔLasB-SEC) drastically downregulated IL-6 (but not IL-8) secretion in F508del-CFTR CFBE; because the levels of trappin-2 in the latter cells were almost undetectable at baseline, the potential further effect of WT-SEC on this mediator was likely not detected in these cells.
Interestingly, WT-SECs had similar effects on IL-6 and trappin-2 secretion in cells from patients with CF (see online supplementary figure 2).
IL-6 up-regulates trappin-2, and LasB disrupts this functional pathway by affecting STAT-3 activation and lung epithelial repair
Having shown that LasB downregulates IL-6 and trappin-2, two anti-inflammatory mediators, we then investigated if there was any functional link between these two molecules. We showed that IL-6 upregulated trappin-2 secretion in NCI-H292 cells (figure 4A) and that WT-SEC (but not ΔLasB-SEC) was able to downregulate this increase (figure 4B).

IL-6 upregulates trappin-2 in NCI-H292 cells, and WT-SEC downregulates IL-6-mediated upregulation of trappin-2 and STAT-3 phosphorylation. Trappin-2 quantification as detected by ELISA in NCI-H292 cell supernatants after treatment with different concentrations of IL-6 (panel A) (n=5, ANOVA) or after a 1 hour pretreatment with IL-6. This was followed by 4 hours treatment with LB, WT-SEC or ΔLasB-SEC (panel B) (n=5, ***p<0.001, two-way ANOVA). Results are shown as mean±SEM. (C) shows a representative Western blot analysis of STAT-3 activation in NCI-H292 cells after 4 hours treatment with LB medium, WT-SEC or ΔLasB-SEC in the presence of IL-1β (1 ng/mL), as detected by antiphospho-STAT3 antibody. Images are representative of three independent experiments. ANOVA, analysis of variance; CFTR, cystic fibrosis transmembrane conductance regulator; IL; interleukin; LB, Luria Broth; SEC, secretomes.
Because IL-6 signalling induces STAT-3 activation23–25 and because some antimicrobial molecules are under the control of this transcription factor,26 27 the effect of LasB was also studied on this pathway. Echoing the results showing that LasB could downregulate IL-6-induced secretion of trappin-2, LasB inhibited STAT-3 activation in NCI-H292 cells (figure 4C).
The IL-6/STAT3 pathway has been shown to be involved in epithelial repair in the lungs and the intestine.23 We showed, in an epithelial scratch assay on NCI-H292 cells, that IL-6 increased epithelial repair (figure 5A) and that WT-SEC (but not ΔLasB-SEC) very significantly hampered this effect (figure 5B). This was also demonstrated in polarised overexpressing WT-CFTR CFBE cells where, in the absence of IL-6, WT-SEC significantly impaired spontaneous repair following cell scratching (figure 5C–D).

LasB decreases basal and IL-6-induced epithelial repair. (A) Epithelial repair, shown as mean±SEM and as measured by scratch assay as the percentage initial (Time 0, (T0) wound 16 hours postscratch in NCI-H292 treated with increasing concentrations of IL-6 (n=4, ANOVA). (B) Effect of WT-SEC and ΔLasB-SEC on basal and IL-6-induced epithelial repair. Injured NCI-H292 cells were pretreated with IL-6 (1 or 10 ng/mL) for 1 hour, and secretomes were added for the following 16 hours (n=5, two-way ANOVA). Results are shown as mean±SEM. Panel C shows representative images, at time 0 and time 16 hours, of the wounded polarised WT-CFTR-CFBE cells that were apically treated with LB, WT-SEC or ΔLasB-SEC. (D) shows the mean±SEM of the percentage of wound closure of three independent experiments of WT-CFTR-CFBE cells apically treated with LB, WT-SEC or ΔLasB-SEC (n=3, ANOVA). ANOVA, analysis of variance; CFTR, cystic fibrosis transmembrane conductance regulator; IL; interleukin; LB, Luria Broth; SEC, secretomes.
LasB is an important P. aeruginosa virulence factor in mice lungs and in vivo transgenic expression of IL-6 and trappin-2 enhances innate immune protection against acute P. aeruginosa infection.
It has been shown in several organ systems that LasB can be very damaging28–30 as reviewed in ref31. Therefore, we evaluated the effect of LasB in our C57BL/6 mouse model. At high doses (≥40 µg), pLasB killed 100% of mice within 5 hours (figure 6A). With a lower dosage of pLasB (5 and 10 µg), significant weight losses were observed 24 hours postintranasal instillation (figure 6B). At that time point, significant lung inflammation and lung injury were apparent in these mice as shown by an increase in cellularity (neutrophils, figure 6C–E), blood (figure 6F) and molecules involved in the host response to infection (figure 6G-J). We then performed ‘rescue experiments’ and showed that Ad-mediated IL-6 (5.108 pfu) overexpression was protective in an acute model of PAO1 (2.107 cfu) infection. Notably, all control PBS and Ad-null-treated C57BL/6 mice died within 50 hours, whereas more than 60% of IL-6 overexpressers survived (statistical significance p=0.01, figure 7A). We reasoned that overexpressing trappin-2 with this low dosage of PAO1WT may not be a significant advantage and we therefore performed a further independent experiment with a higher PAO1WT load to test the effect of trappin-2. In that setting, using the same dosage of Ad vectors (5.108 pfu) and a higher dose of PAO1 (4.108 cfu), we showed that after infection with PAO1, trappin-2 transgenic (eTg) survived significantly longer than C57BL/6 controls (p=0.007), and there was a strong trend (p=0.07) for increased survival in infected eTg mice treated with PBS, compared with infected C57BL/6 controls. Remarkably, trappin-2 and IL-6 double overexpressers had a very significant increased survival over controls and eTg simple overexpressers (all statistically significant, figure 7B).

LasB triggers pulmonary innate immune responses in vivo and is important for pathogenesis. C57BL/6 male mice were intranasally instilled with increasing doses of LasB and survival was monitored (A, numbers in parenthesis represent the number of mice used). Alternatively, PBS (n=3) or moderate doses of LasB (5 µg, n=5 or 10 µg, n=4) or 40 µg were instilled. Mice weight was measured before and 24 hours postinstillation (B). At that time point, mice lungs were recovered and bronchoalveolar lavage (BAL) fluid was analysed for cellularity (C–E) and haemoglobin content (absorbance at 405 nm (F), NB: the original absorbance was multiplied by 10, the BALF dilution factor. In parallel, RNA from lung extracts (homogenised in Trizol) were analysed by qPCR analysis for the expression of TNFα (G), MIP1α (H) and antimicrobial peptides: S100a8 (I) and Lcn2 (J). Results are shown as medians±IQR (n=3–5, ANOVA). ANOVA, analysis of variance.

Overexpression of IL-6 and trappin-2 protects mice in an acute Pseudomonas aeruginosa (PAO1) pneumonia model. (A) C57BL/6 WT mice were instilled intratracheally with either PBS, 5.108 pfu Ad-null or Ad-m-IL-6. Forty-eight hours later, mice were challenged intranasally with a lethal dose (2.107 cfu) of PAO1, and survival was monitored. Statistical significance: C57/B6 + Ad-IL-6 + PAO1 WT versus C57/B6 + Ad-null + PAO1 WT (p=0.0047) and C57/B6 + Ad-IL-6 + PAO1 WT versus C57/B6 + PBS + PAO1 WT (p=0.01). (B) C57BL/6 WT and trappin-2 transgenic mice (C57BL/6 mice expressing human trappin-2, referred as eTg) were instilled as above with either 5.108 pfu Ad-null or Ad-m-IL-6 and were challenged with a higher dose (4.108 cfu) of PAO1, previous to survival monitoring. Statistical significance: eTg + Ad-IL-6+ PAO1 versus C57/B6 + PBS + PAO1 (p<0.0001); eTg + Ad-IL-6+ PAO1 WT versus eTg + PBS + PAO1 (p=0.0038); eTg + Ad-IL-6+ PAO1 WT versus eTg + Ad-null + PAO1 (p=0.0013); eTg + Ad-null + PAO1 versus C57/B6 + PBS + PAO1 (p=0.0070). All data are plotted from the time of PAO1 infection using Kaplan-Meier curves and statistical tests were performed with the GraphPad Prism 6 package, using the Log-rank (Mantel-Cox) test.
In vivo transgenic expression of IL-6 and trappin-2 enhances innate immune protection against acute P. aeruginosa infection by increasing bacterial clearance, decreasing lung injury and engaging tissue repair pathways
Dissecting the mechanisms underlying the phenotype described above, we showed that C57BL/6 mice overexpressing IL-6 (group 2, figure 8A) and eTg mice (transfected or not with Ad-IL-6, group 3 and 4, respectively) had reduced PAO1 loads, when compared with Ad-null WT C57BL/6 infected mice (group 1). Notably, Ad-IL-6 treatment did not further decrease PAO1 load on the eTg background (groups 3 vs 4). This increase in P. aeruginosa clearance was also associated with a clear protection against lung damage, as assessed by measuring haemoglobin content in BALs (OD405nm, figure 8B and C), with the mice most protected being the double IL-6-trappin-2 overexpressers (group 4).

IL-6 and trappin-2 overexpression increase bacterial clearance and decrease lung injury. (A) C57BL/6 WT and eTg mice were instilled as above with either 5.108 pfu Ad-null or Ad-m-IL-6, and 48 hours later challenged (as above) with 106 cfu of PAO1. After a further 24 hours, lungs were recovered, homogenised and used to determine PAO1 cfu count. Results from two independent experiments were pooled (A) and were expressed relative to counts obtained from C57BL/6 mice, which received Ad-null and PAO1 (given a value of 1). Results are medians±IQR (statistical significance: *p<0.005, ANOVA, with each point representing an individual mouse): group 3 versus group 1 (p=0.0047); group 4 versus group 1 (p=0.0003); group 1 versus group 2 (p=0.0003). (B) C57BL/6 WT and eTg mice were instilled as above with either PBS, 5.108 pfu Ad-null or Ad-m-IL-6, and 48 hours later challenged or not (as above) with 1.1.106 cfu of PAO1. After a further 24 hours, BALs were performed and centrifuged. The supernatants were used to assess haemoglobin content by absorbance at 405 nm (top). Results are shown as medians±IQR (statistical significance: *p<0.05, ANOVA, with each point representing an individual mouse): group 1 versus group 2 (p=0.031); group 3 versus group 4 (p=0.0079); group 2 versus group 4 (p=0.031). (C) Tubes containing BALs recovered from mice from groups 1 (C57/Bl6 + Ad-null + PAO1), 2 (C57/Bl6 + Ad-mIL-6 + PAO1), 3 (eTg + Ad-null + PAO1) and 4 (eTg + Ad-mIL-6 + PAO1) are shown for illustrative purposes. ANOVA, analysis of variance; BAL, bronchoalveolar lavage; IL, interleukin.
When BAL cellularity was assessed, Ad-IL-6 treatment showed an ‘anti-inflammatory’ phenotype in the context of PAO1 infection, with a trend towards reduction and increase in neutrophils and lymphocytes, respectively (online supplementary figure 3). In addition, eTg mice treated with Ad-IL-6 showed an increase influx of lymphocytes, compared with WT mice. Furthermore, a transcriptomic study indicated that Ad-IL-6 treatment had an overall downregulatory transcriptional effect in the context of PAO1 infection, in C57BL/6 WT, but above all in eTg mice (online supplementary figure 4).
To further dissect this, we analysed individually by real-time PCR (RT-PCR) a number of genes not represented in the PCR array described above. As expected, we demonstrated, in a non-infectious context, the induction of the IL-6 gene (lower dCT reflecting higher gene induction) following Ad-IL-6 treatment in both C57BL/6 WT and eTg mice (figure 9A). IL-6 was also induced by PAO1 infection and its expression was higher in eTg mice expressing Ad-IL-6 and infected with PAO1 when compared with similarly treated C57BL/6 mice. Trappin-2 expression was, as expected, only detected in eTg mice (since C57BL/6 mice do not express trappin-216 (as reflected here by a dCT>30 in C57BL/6 WT), and PAO1 upregulated its expression in eTg mice (figure 9B). In addition, there was a trend, which did not reach statistical significance, for an increase in trappin-2 expression following Ad-IL-6- (p=0.080) or Ad-IL-6 + PAO1 (p=0.09), respective to Ad-null or Ad-null + PAO1 eTg controls (figure 9B).

IL-6 and trappin-2 overexpression modulate a lung antimicrobial pathway. C57BL/6 WT, and eTg mice were treated and sampled as above (figure 8, (A–B). An aliquot of lung extracts was used for RNA preparation and real-time quantitative PCR (RT-PCR). Results are expressed as dCT=CT gene of interest-CT 18S (house keeping gene). This analysis allows for a non-biased representation, as opposed to the ‘Rq fold increase method’, which requires the choice of a control group, among the 10 different groups analysed. Directly comparable treatments, with differences showing statistical significance are linked together with a horizontal line. Results are shown as medians±IQR (statistical significance: *p<0.05, ANOVA, with each point representing an individual mouse. ANOVA, analysis of variance.
Neither Ad-IL-6 nor trappin-2 overexpression induced KC (figure 9C) or CCL-2 (figure 9D) expression, but as above, PAO1 infection upregulated these cytokines.
PAO1 also induced IL-17 and IL-22 in both C57BL/6 WT and eTg mice. IL-6 + PAO1 treatment was the most potent IL-17 and IL-22 inducer within the eTg group, but also when compared with C57BL/6 WT mice (figure 9E–F).
The existence of an IL-6-IL-17-IL-22-trappin-2 pathway was further demonstrated by showing strong correlations in the expression of these mediators, when all individual mice were analysed together (online supplementary figure 5, panels A–E).
Because the IL-17/IL-22 pathway has been linked to the expression of antimicrobial molecules,32 we tested the expression of other antimicrobials (than trappin-2) in our system. In addition to trappin-2, Ad-IL-6 expression had a clear upregulating effect on Reg-3γ in both WT and eTg mice. In eTg mice receiving Ad-IL-6 and infected with PAO1, there was a (not statistically significant) trend towards increased Reg-3γ expression compared with WT mice treated similarly (figure 9G, p=0.09). Furthermore, Ad-IL-6 and PAO1 strongly increased another antimicrobial, Lcn2, compared with the respective controls, with C57BL/6 and eTg behaving similarly in that respect (figure 9H and online supplementary figure 5 panels F and G).
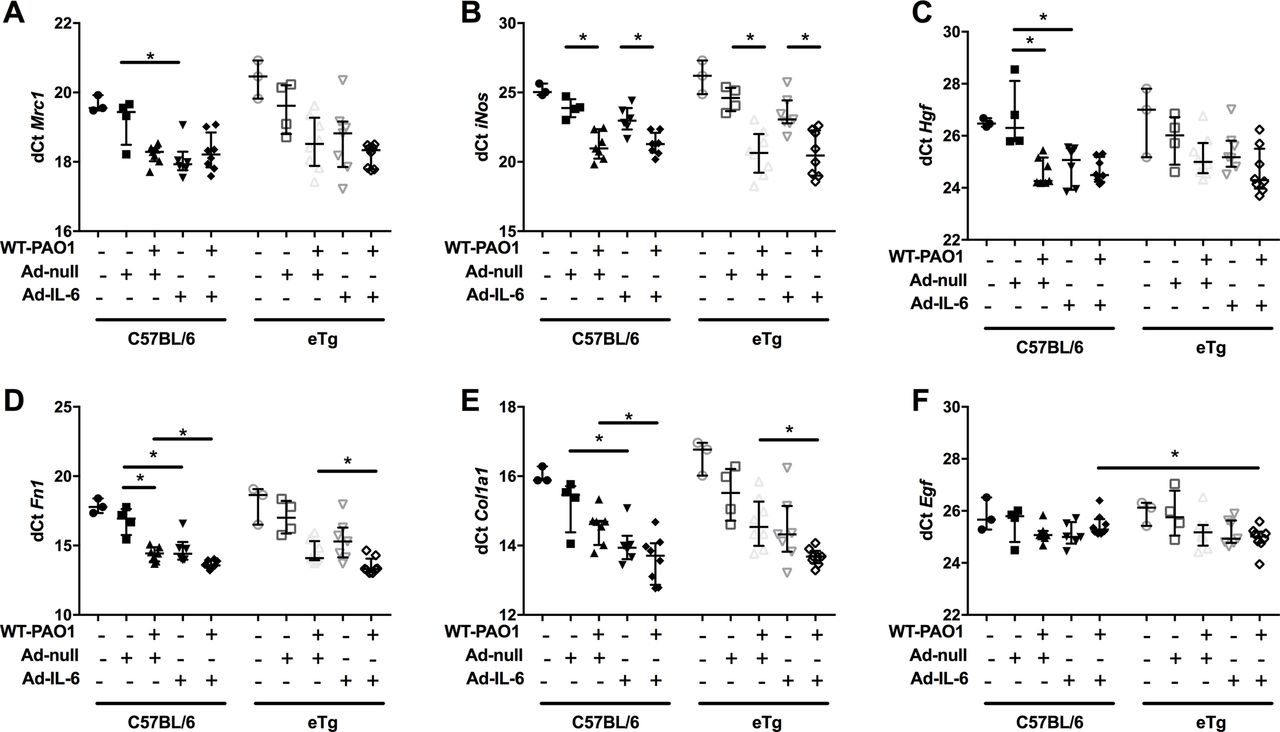
Finally, we showed that overexpression of IL-6 and trappin-2 significantly induced a variety of molecules involved in lung repair. Notably, in C57/Bl6 mice, overexpression of IL-6 increased the expression of Mrc1, Hgf, fibronectin 1 (Fn1) and α1 type I collagen (Col1a1) (figure 10). Furthermore, in the context of PAO1 infection, overexpression of IL-6 and trappin-2 significantly increased mRNA levels of Fn1 and Col1a1. Significant correlations were established between these factors involved in epithelial repair and IL-6 and trappin-2 (online supplementary figure 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
IL-6 and trappin-2 overexpression modulate lung repair molecules gene expression.
Altogether, these experiments (depicted in figures 7–10 and online supplementary figures S3–S6) demonstrate that IL-6 and trappin-2 overexpressers rescue the deleterious effect of WT-PAO1 (LasB expresser) in murine lungs. An outstanding interesting issue will be to assess whether this protective effect also occur in a situation where the less virulent ΔLasB-PAO1 strain infects the lung.
Discussion
P. aeruginosa infections are a common feature of cystic fibrosis and COPD/emphysema exacerbations,14 and we and others have recently shown that the epithelial channel CFTR can be downregulated in vitro and in vivo by exogenous noxious stimuli such as cigarette smoke or endogenous products of innate immune activation, such as neutrophil elastase.6 7 9 33 However, the role of P. aeruginosa and that of its virulence factors on CFTR biology has not been extensively studied.34 35
Among these virulence factors, LasB-bearing P. aeruginosa has previously been shown to be consistently present in mucoid and/or non-mucoid CF sputum secretions.10–13 However, its activity has been studied mainly in vitro where LasB was shown to degrade some cytokines, eukaryotic cell surface receptors and to participate in bacterial escape from the host immune system, in host colonisation and tissue destruction.15 36 37 We show here that LasB is the main protein in the PAO1 secretome (online supplementary figure 1) and that it is the main factor responsible for CFTR degradation in different lung epithelial cell lines, resulting in loss of function. Because CFTR integrity and function is essential to maintain an adequate hydration of mucosae and prevent mucus stasis and bacterial infections (through maintaining adequate acid/base buffering activity and antimicrobial function), the effect of P. aeruginosa LasB on CFTR can be interpreted as a mechanism by which P. aeruginosa breaches the epithelial cell innate immune shield and gains a foothold to colonise the lung of susceptible individuals. Indeed, CFTR degradation by LasB will likely have consequences in CFTR-sufficient patients with COPD but also in CF individuals, including the ones harbouring the F508del-CFTR mutation, since the latter still bears residual CFTR activity (estimated at 5% according to Dekkers et al 38). Similarly, the downregulation of antimicrobial molecules such as trappin-2 shown here is likely to be beneficial for the bacterium. We show here, in epithelial cell lines and primary cells from patients with CF, that this is associated with a drastic downregulation of IL-6 (at both transcriptional and translational levels, not shown) but that IL-8 levels are not affected.
Although other P. aeruginosa PAMPs such as LPS and, to a lesser extent, flagellin, have been used extensively to model P. aeruginosa infection in vivo, surprisingly, the direct effect of LasB has been, so far, clearly understudied, and little is known about its effects in the lung.31 39–41 Here, we showed LasB to be inflammatory at moderate doses (5–10 µg), by inducing lung damage (figure 8E), neutrophilic recruitment (figure 8D) and by transcriptionally upregulating proinflammatory cytokines and antimicrobial molecules (figure 8F-I). At higher doses (40 µg), LasB was shown to be lethal (figure 8A). In that context, echoing the in vitro data demonstrating the protective effect of IL-6 and trappin-2 following LasB exposure, we show that transgenic expression of IL-6 and trappin-2 enhanced protection against WT PAO1, a P. aeruginosa strain expressing LasB.42 Remarkably, all control mice died, whereas a significant proportion survived with either IL-6 or trappin-2 transgenic expression. Surprisingly, although IL-6 has been shown previously to be associated in vivo with reduced local lung and systemic inflammatory responses, following LPS pulmonary and intra-peritoneal instillation, respectively,43 this is, to our knowledge, the first report demonstrating the beneficial effect of IL-6 in any pneumonia model. Indeed, IL-6 is not classically described as a molecule with ‘direct’ anti-antimicrobial properties, and the latter may not fully account on its own for the results obtained here. It seems more likely that IL-6 acts through the induction of a variety of mediators (IL-17, IL-22, antimicrobials and molecules implicated in tissue repair, confirming in vitro data), which overall dissuade neutrophilia (no change in KC was observed here) and promote lymphocytic influx and tissue repair, as observed in other systems.23–25 44–46 Furthermore, although trappin-2 itself 47 48 and defensins49 have been shown to be associated with repair processes (although mostly in other organs, and with little mechanistic information), we show here clearly, as stated above, that the IL-6 + trappin-2 dual regimen was the most effective in protecting mice against P. aeruginosa-induced pneumonia (figures 8 and 9). This suggests the existence of a synergy between these two mediators, both engaging anti-inflammatory/prorepair pathways, and ultimately leading to faster lung healing and removal of the microbial burden, improving tissue resilience and remodelling (figure 10; online supplementary figure 6).
Finally, and demonstrating the potential clinical relevance of the described IL-6-antimicrobial pathway, an additional interesting finding from our study is that, aside from the LasB effect, the levels of IL-6 and trappin-2 (but again not those of IL-8) were drastically reduced in mutant F508del-CFTR CFBE cells, compared with WT-CFTR-corrected CFBE cells. This advocates that the inhibition of the IL-6-trappin-2 pathway described here may be operative in cystic fibrosis. However, this will need to be confirmed in vivo since overexpression of CFTR in air liquid interface (ALI)-differentiated epithelial cells from four different F508del-CFTR individuals had no effect on the protein recovery of trappin-2, IL-6 and IL-8, when compared with Ad-null-infected control cells. Nonetheless, this could be explained by the short restoration of CFTR expression (48 hours treatment), which may not be optimal to modulate the F508del phenotype and restore the potentially deficient IL-6-trappin-2 pathway.
Regardless, a tantalising interpretation of our data could be that LasB, one of the major (and consistently found in CF secretions10–13) virulence factors of P. aeruginosa: (1) targets CFTR and, in doing so, modifies the lung mucosal milieu leading to ionic flux disturbances, increased acidity and antimicrobial loss of function; (2) targets trappin-2, an important antibacterial/anti-inflammatory molecule; (3) hampers an IL-6/repair pathway while leaving untouched a ‘chronic neutrophilic program’, contributing to overzealous inflammation and lung damage.
Our novel findings hold important implications for both genetically CFTR-sufficient individuals infected with P. aeruginosa (eg, during nosocomial infections, or during COPD exacerbations), as well as for individuals with CF. Directly targeting the virulence factor LasB or upregulating the downstream host immune molecules IL-6 and trappin-2 represents new therapeutic strategies for such patients.
Acknowledgments
We wish to thank Brigitte Solhonne for technical assistance.
References
Footnotes
Contributors VS-C designed and performed experiments, analysed data and wrote part of the manuscript. BV, FB and SK performed ex vivo and in vivo experiments. AH helped perform ex vivo experiments. AC blindly performed histological analysis. ZX provided adenovirus constructs and critically appraised drafts of the document. IS-G and AE provided CF patients bronchial epithelial cells and wrote a section of the manuscript. IG-V helped in the design of the experiments and critically appraised drafts of the document. J-MS designed experiments, provided reagents, analysed data and wrote the manuscript.
Funding ‘Vaincre la Mucoviscidose’ (grants RF 20130500896 and RF 20150501368).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves