Article Text

Abstract
Rationale In infants and young children with cystic fibrosis, lower airway infection and inflammation are associated with adverse respiratory outcomes. However, the role of lower airway microbiota in the pathogenesis of early cystic fibrosis lung disease remains uncertain.
Objectives To assess the development of the lower airway microbiota over time in infants and young children with cystic fibrosis, and to explore its association with airway inflammation and pulmonary function at age 6 years.
Methods Serial, semi-annual bronchoscopies and bronchoalveolar lavage (BAL) procedures were performed in infants newly diagnosed with cystic fibrosis following newborn screening. Quantitative microbiological cultures and inflammatory marker (interleukin 8 and neutrophil elastase) measurements were undertaken contemporaneously. 16S ribosomal RNA gene sequencing was conducted on stored BAL samples. Spirometry results recorded at 6 years of age were extracted from medical records.
Measurements and main results Ninety-five BAL samples provided 16S ribosomal RNA gene data. These were collected from 48 subjects aged 1.2–78.3 months, including longitudinal samples from 27 subjects and 13 before age 6 months. The lower airway microbiota varied, but diversity decreased with advancing age. Detection of recognised cystic fibrosis bacterial pathogens was associated with reduced microbial diversity and greater lower airway inflammation. There was no association between the lower airway microbiota and pulmonary function at age 6 years.
Conclusions In infants with cystic fibrosis, the lower airway microbiota is dynamic. Dominance of the microbiota by recognised cystic fibrosis bacterial pathogens is associated with increased lower airway inflammation, however early microbial diversity is not associated with pulmonary function at 6 years of age.
- Cystic Fibrosis
- Bacterial Infection
- Bronchoscopy
- Paediatric Lung Disaese
Statistics from Altmetric.com
Key messages
What is the key question?
How does the lower airway microbiota in infants and young children with cystic fibrosis contribute to disease progression?
What is the bottom line?
The diversity of the lower airway microbiota in young children with cystic fibrosis decreases with age, and the presence of respiratory bacterial pathogens is associated with reduced microbial diversity and increased airway inflammation.
Why read on?
This study explored the relationship between the early evolving lower airway microbiota, accompanying airway inflammation, and later pulmonary function in infants and children with cystic fibrosis.
Introduction
Presymptomatic infants with cystic fibrosis (CF) diagnosed following newborn screening can already have evidence of infection and inflammation in their lower airway samples. These changes are associated with earlier development of bronchiectasis and worse pulmonary function later in childhood.1 ,2 Particular pathogens, especially Staphylococcus aureus and Pseudomonas aeruginosa, are associated with increased airway inflammation.3 ,4 Recent molecular techniques have identified the complex, polymicrobial nature of CF lung disease, with obligate and facultative anaerobes recovered increasingly from the airways.5–7 However, little is known about the role of these communities in the pathogenesis of CF lung disease.
Organisms comprising the human microbiota have complex relationships with one another and their host, where resident bacteria have potentially site-specific roles in immune modulation and response to individual pathogens.8 Indeed, the upper and lower airways are distinct anatomical compartments with different microbial profiles. In health, the lower airways are not sterile, and while the lower airway microbiota of healthy children is poorly described, it is postulated that in disease states microbial diversity is altered, as either a primary or a secondary phenomenon, by colonisation with proliferating cells and by suppression of innate immunity and airway clearance.8
The airway microbiota is linked to progressive CF lung disease. Cross-sectional studies of adults and older children with CF suggest a relatively diverse lower airway microbiota,9 ,10 with decreased microbial diversity in older patients correlated with severe lung disease.6 ,7 ,10–12 Analyses of explanted lung tissue suggest markedly reduced microbial diversity in advanced disease states dominated by recognised CF pathogens, such as S. aureus, P. aeruginosa and Burkholderia cepacia complex.7 ,13 ,14 Evaluating the early lower airway microbiome and its relationship to pulmonary inflammation is essential to understanding the pathogenesis and therefore potentially altering the course of CF lung disease. Here, we describe the early lower airway microbiota in children with CF and explore its relationship with lower airway inflammation and pulmonary function at age 6 years. We hypothesised that reduced bacterial diversity during early childhood is associated with lower lung function.
Methods
Participants
Children identified with CF following newborn screening or as neonates with meconium ileus and sweat chloride concentrations >60 mmol/L were recruited into a single-centre study in Melbourne, Australia from 1992 to 2001. As reported previously, bronchoscopy and bronchoalveolar lavage (BAL) were performed at the time of study entry, annually thereafter, and during hospitalisation for pulmonary exacerbation.15–17 BAL samples were obtained from subjects' right middle lobe and lingula bronchi, and except for a small subset,16 these aliquots were pooled at the time of collection. Inflammatory markers, interleukin 8 (IL-8) and neutrophil elastase (NE) were measured and quantitative microbiological culture performed contemporaneously, as described previously.15 ,16 NE ≥5 μg/mL was considered elevated, consistent with assay detection limits.15 Whole BAL samples were stored at –70°C. 16S ribosomal RNA gene (16S rRNA gene) analysis was performed on available samples. Subjects with at least one BAL sample and corresponding 16S rRNA gene data were included in this study. The Royal Children's Hospital Melbourne Human Research Ethics Committee approved the study (DA001-2015-32).
Clinical data
Demographic information was obtained at enrolment. Clinical data, including respiratory symptoms (cough, respiratory hospitalisation) and antibiotic use (oral, inhaled or intravenous) were recorded at 3-monthly clinical assessments and at bronchoscopy. The best FEV1 and FVC at age 6 years were obtained from medical records; per cent predicted values were recalculated using Global Lung Initiative 2012 reference values.18
16S rRNA gene sequencing
Total nucleic acid was extracted from 200 µL of whole BAL fluid for 16S rRNA sequence analysis. EasyMAG Lysis Buffer was added to a total volume of 2 mL, and cells were disrupted by bead beating with 0.1 mm MO BIO glass beads (catalogue number 2600-50B2) using the MO BIO vortex adapter at 4°C for 10 min. Beads were pelleted in a microcentrifuge at maximum speed for 1 min, and the supernatant was removed and transferred to the NucliSENS easyMAG instrument (bioMérieux, Inc, Marcy l'Etoile, France). Nucleic acid extraction followed the manufacturer's protocol using off-board lysis. Total nucleic acid was eluted in 50 µL easyMAG Extraction Buffer 3 and 16S rRNA gene sequences were generated on the Roche 454 platform, using the V1-3 variable regions following Human Microbiome Project protocols.19 In brief, the V1–3 region was amplified using the following primers, adding index sequences to distinguish each sample: 27F–5′ CCTATCCCCTGTGTGCCTTGGCAGTCTCAG_AGAGTTTGATCCTGGCTCAG; 534R–5′ CCATCTCATCCCTGCGTGTCTCCGACTCAG_INDEX_ATTACCGCGGCTGCTGG. The cycling conditions were: 95°C, 2 min; 30 cycles each at 95°C, 20 s; 56°C, 30 s; 72°C, 5 min; after cycling held at 4°C. PCR products were cleaned using Agencourt AMPure Beads (Beckman Coulter, Indianapolis, Indiana, USA), and DNA concentrations were determined using the Qubit Fluorometer (ThermoFisher, Waltham, Massachusetts, USA). Samples were pooled in equimolar concentrations, except for negative controls and poorly amplified samples, which were added in their entirety. The pools were concentrated using a Qiagen MinElute column following the manufacturer's instructions (Qiagen, Germantown, Maryland, USA) and eluted in 30 µL 1× low TE buffer, pH 8.0. Sequences were generated on the Roche 454 GS FLX Titanium Instrument (Roche/454 Life Sciences, Branford, Connecticut, USA). Sequences were binned, based on the index sequences, allowing one mismatch. Low-quality reads were removed when average quality scores were <Q35 or the read length <200 base pairs. Chimeric sequences were identified using NAST-iEr and Chimera Slayer,20 and chimeric sequences were removed. Reads passing quality control were classified using Ribosomal Database Project naïve Bayesian classifier,21 V.2.2 with training set 6. Sequences were classified to the lowest taxonomic level that could be assigned with confidence values >0.5. Negative control samples from PCR reagents and the buffer processed with the extraction protocol in parallel with the samples were included, and these yielded 1–359 sequence reads.
Statistical analysis
Sequence data from patient samples were subsampled to 1000 reads for analysis of community structure,22–24 and were correlated with culture and inflammatory data from the corresponding lobe. Sequences not classified to phylum level were removed before analysis. The relative abundance of sequences assigned to genus level was calculated from each BAL sample; individual genera that accounted for ≥60% of all sequences in a single sample were defined as ‘dominant’. The Shannon Diversity Index (SDI) of individual samples was calculated using 1000 random sequences from each sample. This was repeated 100 times and the mean value reported. Study data were managed using REDCap electronic data capture tools hosted at Murdoch Children's Research Institute.25 Statistical analysis was performed using ‘R’ (R Foundation for Statistical Computing, Vienna, Austria). IL-8 and NE values were logarithmically transformed (log10) for analysis because of their skewed distributions. When NE activity was below the assay detection threshold, a value of 2.5 μg/mL was assigned. Generalised estimating equations regression with unstructured correlation and robust SEs, linked by subject identifier, were used to account for multiple samples from the same subject. The regression model used Gaussian with identity link and binomial distribution with logit link for continuous and categorical variables respectively. χ2, paired t tests and linear regression were used if appropriate. β diversity was assessed using Bray-Curtis dissimilarity.
Results
Subjects
The 16S rRNA V1–3 region could be amplified and sequenced for bacterial community analysis in 95 samples from 48 subjects, including serial longitudinal samples from 27 subjects. At least 1000 reads were generated from each of these samples. Amplification failed or was poor in an additional 53 samples, possibly due to low bacterial load and/or sample age (see online supplementary data). Subjects' characteristics are presented in table 1, and except for p.Phe508del homozygosity, these did not differ significantly from the remaining birth cohort at recruitment (table 1). CF was diagnosed early, at a median age of 1.4 months (IQR 1.0–1.7-months). Samples collected from those with respiratory symptoms were more likely to yield ≥1000 sequences (see online supplementary table S1).
Demographic details of subjects with BAL samples included for analysis compared with remainder of original birth cohort at recruitment
supplementary data
Samples were obtained between 1.2 and 78.3 months of age (median age 20.4 months, IQR 13.0–32.1 months). Two samples were collected from 9 subjects, three samples from 16 subjects, and four samples from 2 subjects (see online supplementary figure S1). Fifty-four (63%) and 45 (51%) samples were obtained from subjects with respiratory symptoms and those receiving antibiotics respectively (table 2). Eleven (13%) samples were collected during respiratory hospitalisation. Thirty samples were obtained from subjects without respiratory symptoms and not taking antibiotics at the time of BAL. Subjects reporting respiratory symptoms were significantly older and more likely to be taking antibiotics (see online supplementary table S2). Seventy-eight (82%) samples were pooled from the right middle lobe and lingula, and 11 (12%) and six (6%) were separate samples from the right middle lobe and lingula respectively.
Bronchoalveolar lavage (BAL) samples (n=95) from which the 16S rRNA V1–3 region could be amplified and sequenced for bacterial community analysis
Lower airway microbiota in young children with CF
Overall, 1 201 177 individual sequences were identified, 98% of which could be classified to genus level. The most prevalent phyla were Firmicutes (63.7%), Proteobacteria (21.9%), Actinobacteria (8.1%), Bacteroidetes (4.8%) and Fusobacteria (1.3%). Staphylococcus was the most prevalent genus (36.6%), followed by Streptococcus (13.7%), Pseudomonas (6.1%), Neisseria (5.4%), Haemophilus (4.9%), Gemella (4.9%), Graniculatella (3.9%), Prevotella (2.8%), Veillonella (2.6%) and Streptomyces (2.1%) (figure 1).
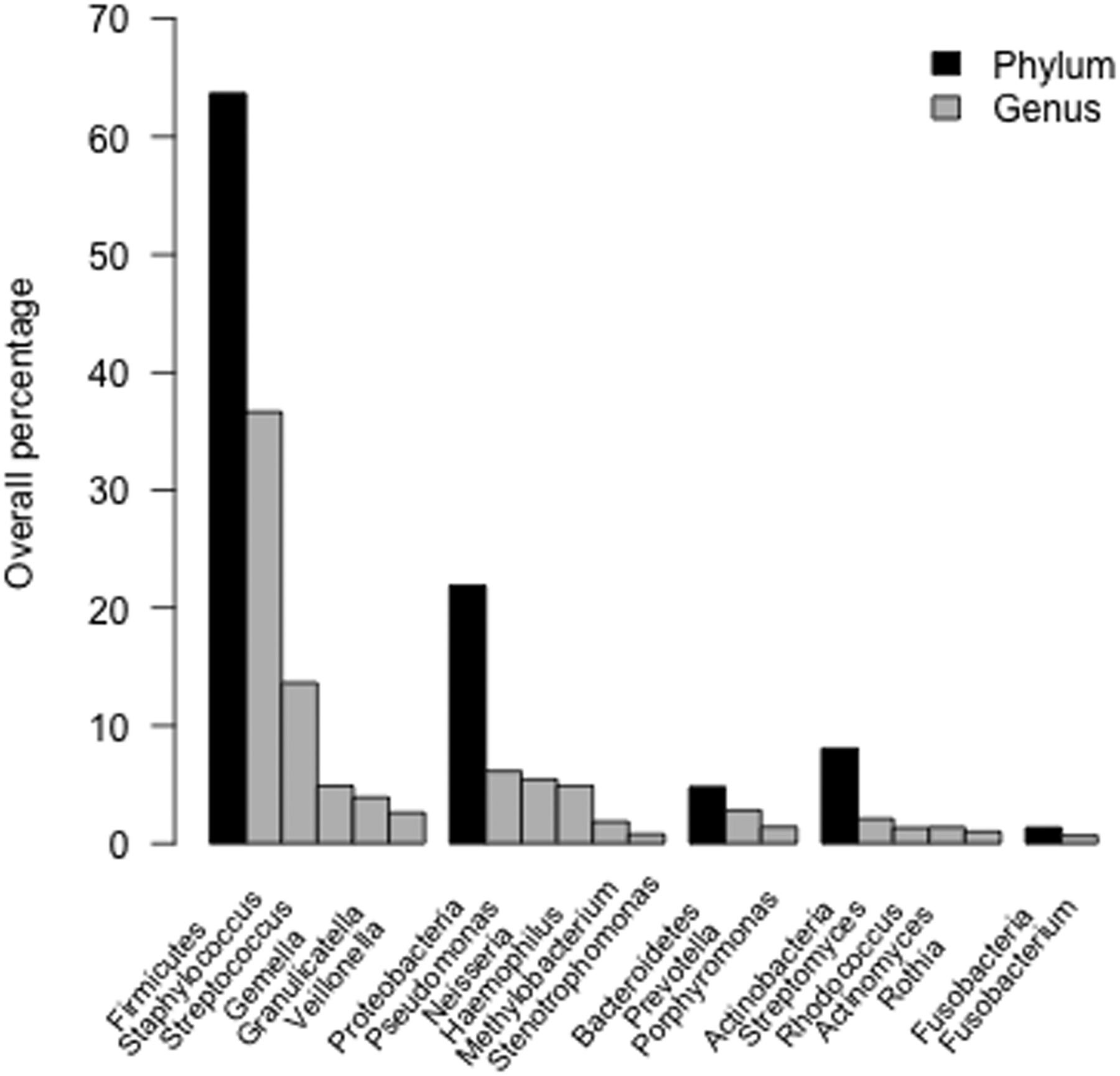
Prevalence of phyla and genera. Sequences were classified to genus level. The overall percentage of reads assigned to individual phyla and genera are displayed. The genera depicted account for 91.6% of sequences detected in the bronchoalveolar lavage samples. The relative abundance values in the respective phylum categories not accounted for by the genera presented are: Firmicutes 1.97%, Proteobacteria 2.74%, Bacteroidetes 0.56%, Actinobacteria 2.23% and Fusobacteria 0.68%.
When genera corresponding with recognised CF pathogens were detected on 16S rRNA gene analysis, they were occasionally dominant, accounting for ≥60% of all reads in that sample (figure 2). Staphylococcus was detected in 69 (73%) samples and was the dominant genus in 18 (26%). Pseudomonas was detected in 62 (65%) samples and was dominant in 5 (8%). Stenotrophomonas and Burkholderia were detected in 20 (21%) and 18 (19%) samples respectively and each was dominant in one. Haemophilus was detected in 59 (62%) samples, but was the dominant genus in just 2 (3%). Both of these latter samples yielded Haemophilus influenzae in quantitative culture.
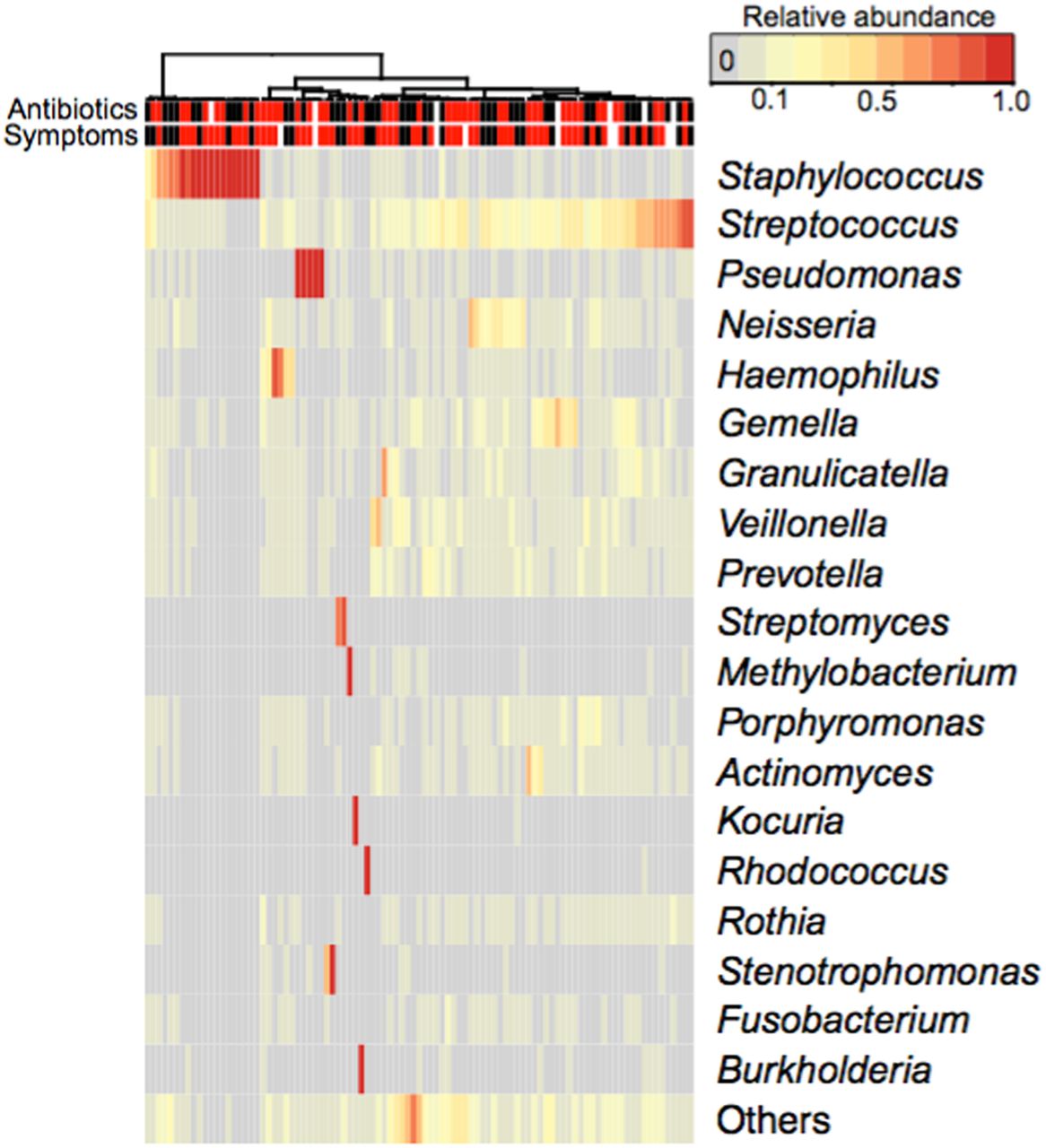
Relative abundance of individual genera at each sampling time. Individual genera with relative abundance >0.5% are depicted in rows. Individual bronchoalveolar lavage (BAL) samples are displayed in columns. Samples are organised according to similarity. The coloured, vertical bars at the top indicate antibiotic treatment and respiratory symptoms at the time of BAL (red—yes, black—no, white—unknown). The relative abundance of each genus in each BAL sample is reflected in the intensity of the heat map colour.
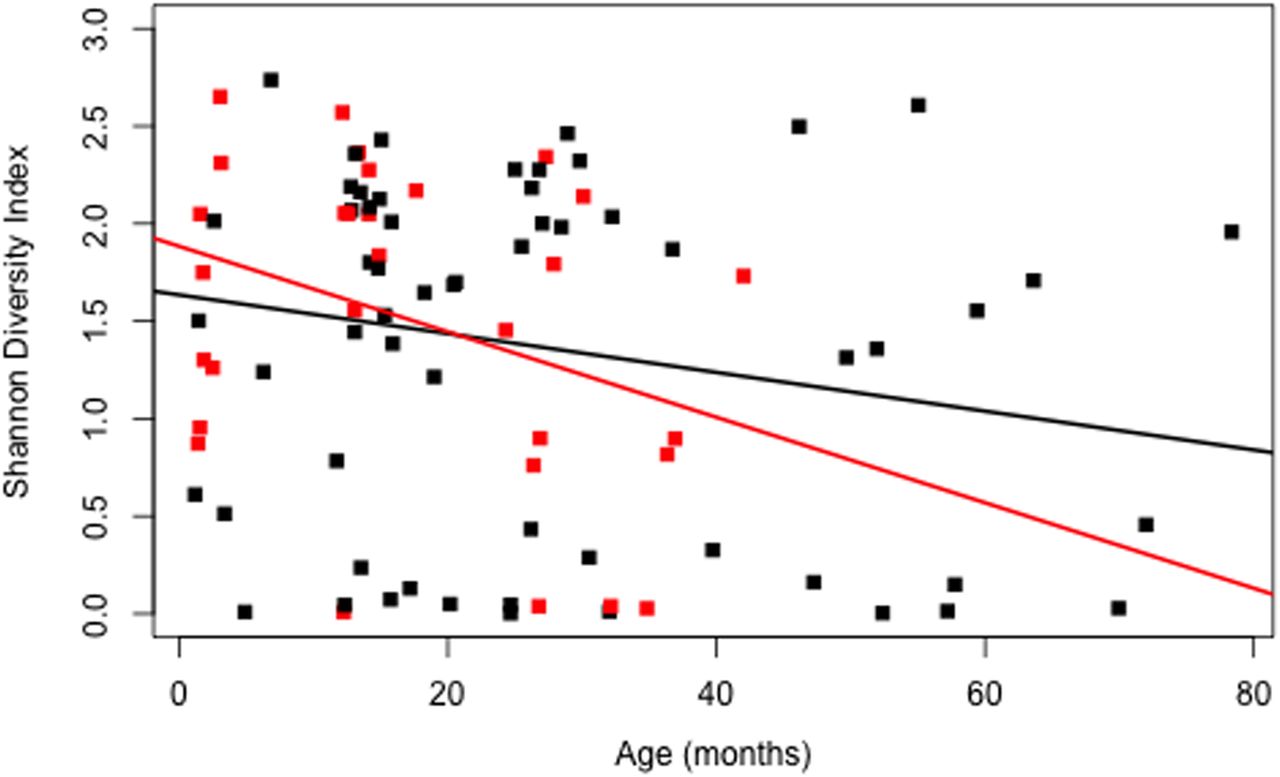
Although considerable variability existed between individuals (see online supplementary figure S2), overall microbial diversity decreased in serial BAL samples in association with increased patient age (β=−0.01, p=0.03), including in the absence of respiratory symptoms and antibiotic use (β=−0.02, p=0.02, figure 3). After adjustment for age and antibiotic use, the presence of respiratory symptoms was associated with reduced microbial diversity (β=−0.35, p=0.02). There was no association between microbial diversity and CF transmembrane regulator mutation (β=−0.2; p=0.16), respiratory symptoms (β=−0.27, p=0.20) or antibiotic use (β=−0.002; p=0.97) at bronchoscopy.
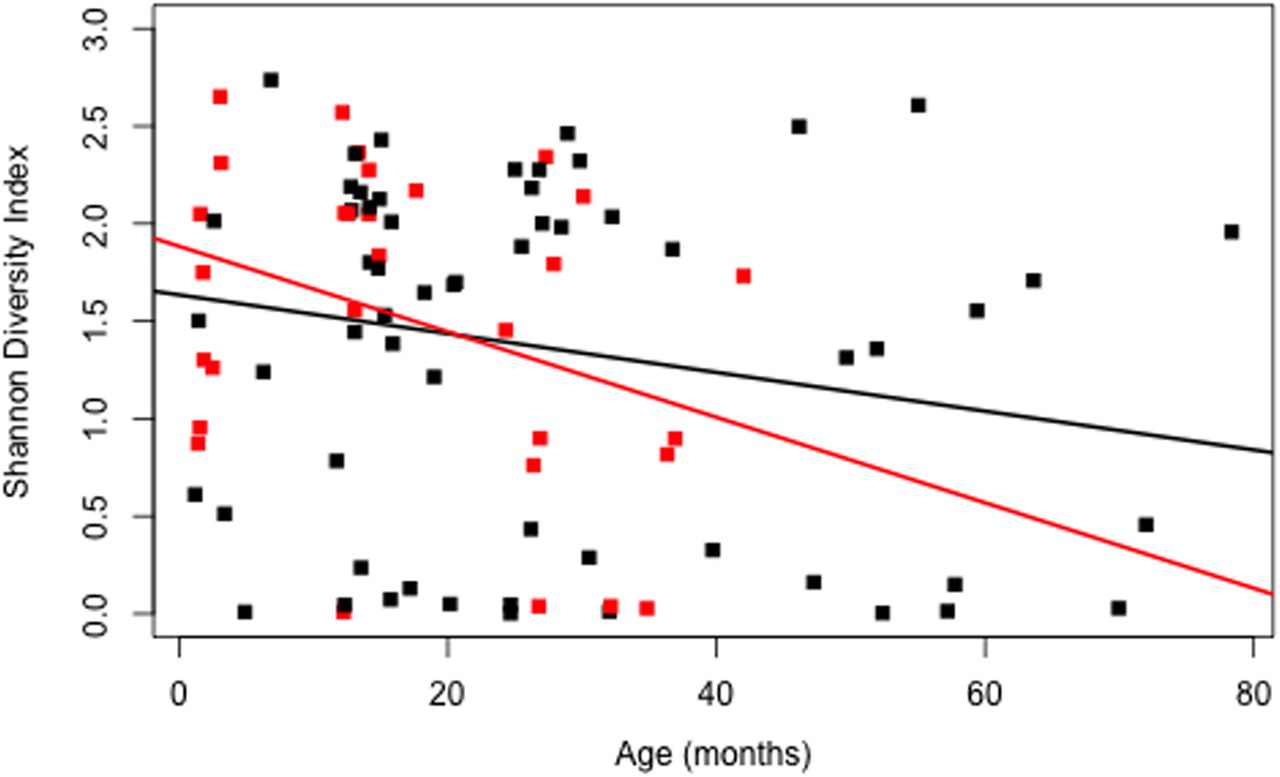
Shannon Diversity Index versus subject age (months) (β=−0.01, p=0.031). Samples obtained during clinical stability, in the absence of respiratory symptoms or antibiotic use, are depicted in red (β=−0.02, p=0.02).
Correlation of the lung microbiota with quantitative culture
16S rRNA gene analysis confirmed the finding of recognised CF pathogens by culture. In all samples where S. aureus or P. aeruginosa grew in concentrations ≥105 colony forming units (cfu)/mL, Staphylococcus or Pseudomonas respectively were detected in 16S rRNA gene analysis. S. aureus was detected by quantitative culture at concentrations ≥105 cfu/mL in seven BAL samples. In five of the seven samples, Staphylococcus was the dominant genus in 16S rRNA gene analysis. Staphylococcus was dominant in 16S rRNA gene analysis of 13 additional samples; in 11 of these, S. aureus was also detected by culture. Similarly, Pseudomonas was dominant in 16S rRNA gene analysis of five BAL samples; in all five, P. aeruginosa cultures yielded ≥105 cfu/mL. P. aeruginosa was also present in concentrations ≥105 cfu/mL in two additional samples; in both, Pseudomonas was detected, but not dominant on 16S rRNA gene analysis. B. cepacia complex and Stenotrophomonas maltophilia each grew in concentrations ≥105 cfu/mL in a single BAL sample. In 16S rRNA gene analysis of the respective samples, Burkholderia was dominant and Stenotrophomonas was detected in a relative abundance of 59.6%. In the only BAL dominated by Stenotrophomonas, S. maltophilia grew in a concentration of 102 cfu/mL. Cultured organisms regarded as upper respiratory commensals (eg. α-haemolytic streptococci and Neisseria species) were supported by 16S rRNA gene analysis findings of corresponding genera in variable abundance (table 3). Furthermore, 16S rRNA gene analysis frequently revealed the presence of organisms that went undetected by routine culture.
Agreement between microbiological culture and 16S rRNA gene analysis
Lower airway inflammation and the lung microbiota
Dominance of established proinflammatory organisms (Pseudomonas, Staphylococcus, Streptococcus, Haemophilus, Stenotrophomonas and Burkholderia)3 on 16S rRNA gene analysis was associated with higher inflammation indices in the lower airway (figure 4). In BAL samples with a dominant pathogen genus, the median IL-8 and NE were 1041 (IQR 210–2252) pg/mL and 18.4 (IQR 6.0–37.8) μg/mL respectively, compared with 323 (IQR 84–875) pg/mL and 5.7 (IQR 2.5–14.4) μg/mL in other samples (IL-8, p=0.002; p=0.0008). IL-8 and NE concentrations in BAL samples dominated by Streptococcus were comparable to those without a dominant pathogen genus (IL-8, median 254.5 pg/mL, IQR 136–765 pg/mL; NE, median 3.9 μg/mL, IQR 2.5–6.8 μg/mL) (figure 4).
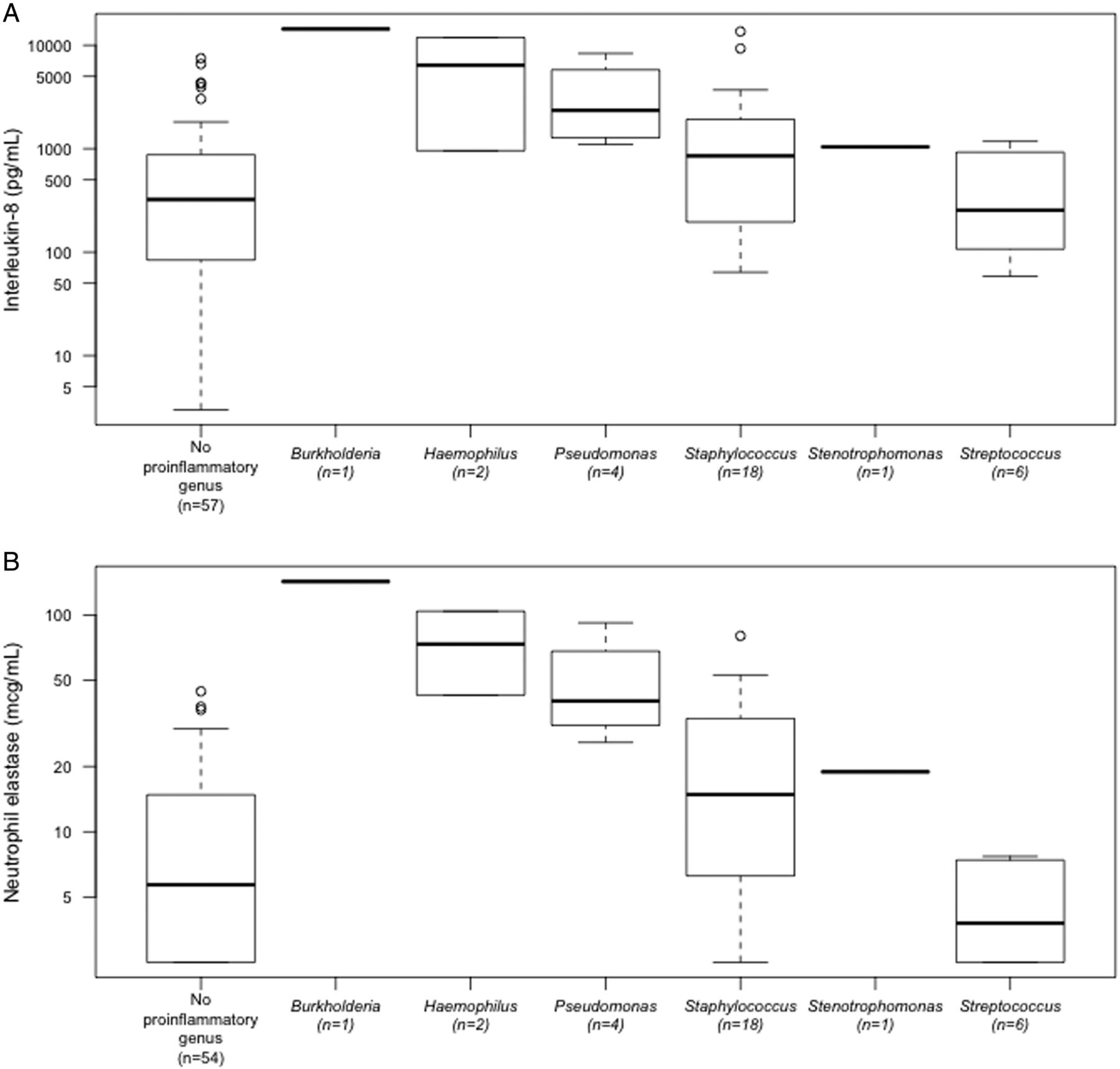
Dominant proinflammatory organisms by 16S rRNA gene analysis and (A) interleukin 8 (IL-8) and (B) neutrophil elastase (NE) concentrations. Data for bronchoalveolar lavage samples with corresponding IL-8 (n=89, 94%) and NE (n=86, 91%) are displayed.
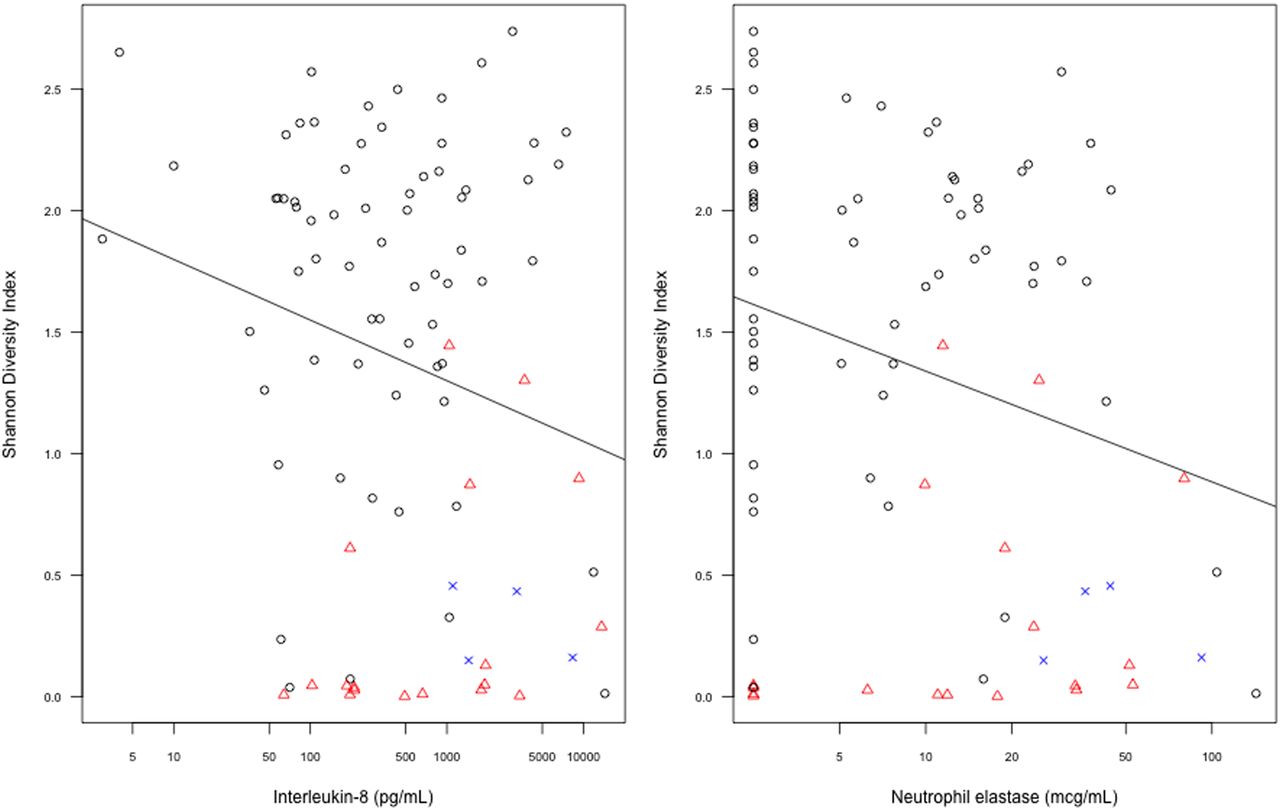
Reduced microbial diversity was associated with increased BAL NE (β=−0.5, p=0.02, age-adjusted β=−0.43, p=0.03), although not IL-8 (β=−0.23, p=0.05, age-adjusted β=−0.17, p=0.097). Figure 5 illustrates the relationship of microbial diversity, IL-8 levels, NE concentrations, and dominance of pathogen genera, Staphylococcus and Pseudomonas.

Interleukin-8 and neutrophil elastase concentrations versus the Shannon Diversity Index (age adjusted, p=0.097 and p=0.03 for interleukin 8 and neutrophil elastase respectively). Samples dominated by Staphylococcus and Pseudomonas are shown in red and blue respectively.
Compared with asymptomatic subjects, children symptomatic at the time of bronchoscopy had increased lower airway inflammatory markers (IL-8: median 864 (IQR 196–1833) vs 195 (IQR 68–547) pg/mL, t-test log10 IL-8, p=0.005; NE: median 13.0 (IQR 3.6–25.3) vs 5.8 (IQR 2.5–12.0) μg/mL, t-test log10 NE, p=0.02). In subjects who had bronchoscopies when both symptomatic and asymptomatic, symptoms were associated with a trend towards increased inflammation (IL-8 (n=14): mean of differences, 1343 pg/mL, 95% CI −888.2 to 3574.2 pg/mL, p=0.2; NE (n=13): mean of differences, 16.3 μg/mL, 95% CI −2.6 to 35.5, p=0.08). A dominant proinflammatory genus was detected in 22/54 (41%) samples from symptomatic subjects and in 8/31 (26%) samples from those without symptoms (p=0.17).
Change in microbiota over time
The composition of individuals' lower airway microbial communities varied considerably over time (see online supplementary figure S2). The β diversity of serial samples obtained from individual subjects was comparable to the β diversity of samples obtained from different subjects (median Bray-Curtis dissimilarity (IQR): 0.91 (0.7–0.99) and 0.9 (0.72–0.99) respectively) with the exception of samples dominated by known proinflammatory genera (figure 6).
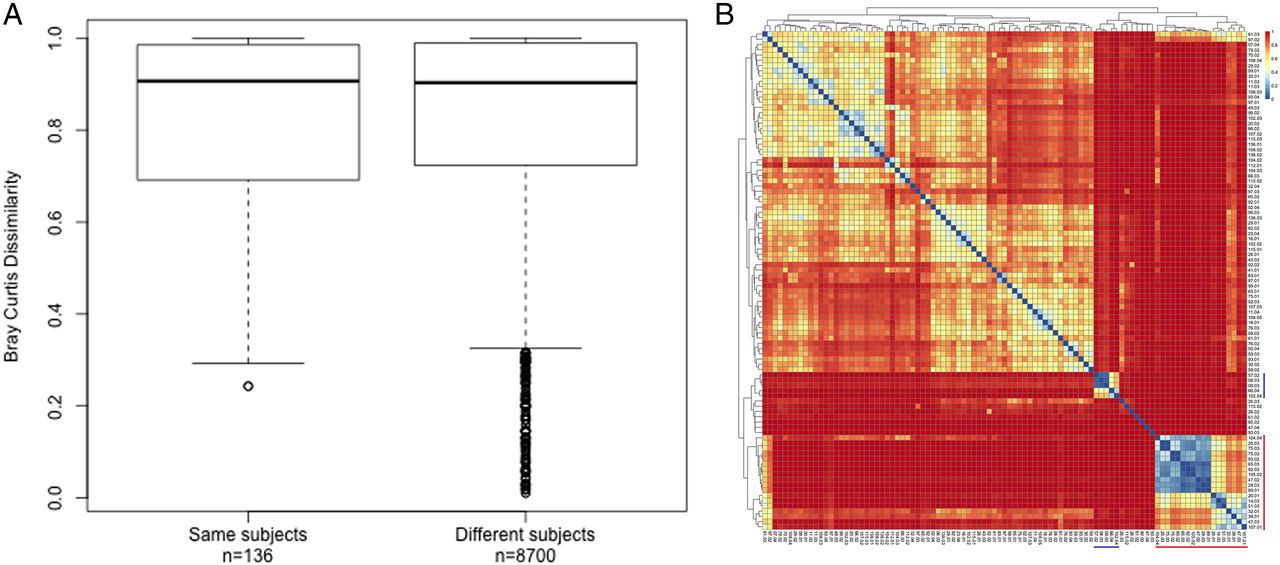
Bray-Curtis dissimilarity of bronchoalveolar lavage (BAL) samples. (A) Box plot summarising the pairwise Bray-Curtis dissimilarity of BAL samples obtained from the same and different subjects, and (B) heatmap of all pairwise distances. Outliers in (A) correlate with pairs of samples dominated by known proinflammatory pathogens.
Pulmonary function measurements and microbiota
There was no relationship between subjects' bacterial diversity and their best pulmonary function measures at age 6 years (figure 7). Neither lowest SDI nor reduced early microbial diversity (age <6-months) was associated with pulmonary function measures at age 6 years (r2=0.03, p=0.26 and r2=0.001, p=0.9 respectively).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Shannon Diversity Index and best FEV1 at 6 years of age. Individual subjects' with available spirometry data are displayed on the horizontal axis (n=42, 88%); their FEV1 (percentage predicted) and Shannon Diversity Index (•) are displayed on the vertical axes.
Discussion
We examined the evolution of lower airway microbiota in young children with CF over time using BAL samples that were collected semi-annually up to 24 years ago at the Royal Children's Hospital Melbourne, and its association with airway inflammation and pulmonary function at age 6 years. The lower airway microbiota was diverse, but diversity decreased with subject age. Genera of known CF pathogens often emerged, and their dominance was associated with lower airway inflammation. We were unable to identify any relationship between microbial diversity and pulmonary function at age 6 years.
To our knowledge this is the first longitudinal molecular study of BAL samples from infants with CF and provides unique insights into their lower airway microbiota during early childhood. Recruitment from a single birth cohort not routinely exposed to prophylactic antibiotics and inclusion of long-term outcome data add to the study's ability to provide information about CF lung disease progression and its relationship to the lower airway microbiota. The study does however have important limitations. It involves the secondary analysis of prospectively collected BAL samples from two decades ago. Rigorous storage and handling practices and strict laboratory exclusion criteria were implemented to ensure the quality of the 16S rRNA gene data. These data correlated with the participants' own culture results, assessed contemporaneously from the same BAL samples, and previous findings from lower airway samples in older patient cohorts,26 and indicate that they accurately represent the subjects' lower airway microbiota. In order to address the potential for reagent contamination, negative sequence controls were processed alongside study samples and only the 64% of samples that could be subsampled to 1000 reads were included in analysis of microbial community structure. Further methodological studies are required to explore the molecular analysis of BAL fluid from infants and very young children, who may have both very low sample volumes and total bacterial load. Future prospective, longitudinal studies of BAL fluid are also required to determine the impact of cumulative antibiotic exposure on the airway microbiota. Strict procedural protocols were maintained to minimise potential contamination with upper airway secretions. Nevertheless, it is not possible to fully differentiate between upper airway organisms introduced at the time of bronchoscopy and the resident lung microbiota.
The lower airway microbiota was notably diverse in our cohort of newly diagnosed infants with CF. The most prevalent phylum was Firmicutes, and Staphylococcus the most prevalent genus. The facultative anaerobes now frequently detected in molecular analyses of sputum specimens from adults with CF, Gemella, Prevotella and Veillonella, represented 4.9%, 2.8% and 2.6% of overall genera respectively. Interestingly, a recent comparison of BAL samples from 13 older children with CF (mean age 3.95 years, range 1.1–8.1 years) and nine controls (mean age 3.78 years, range 0.9–9.75 years), similarly showed reduced diversity in the CF population.27
The association between reduced diversity with increasing subject age is consistent with findings from longitudinal studies of lower airway samples from adults with CF.10 ,11 It does, however, contrast with the findings of previous longitudinal analyses of children with CF, which only examined the upper airway microbiota.10 ,28–32 During infancy, the nasopharyngeal microbiota is dynamic,33–35 although differences between that of infants with CF and healthy controls have been reported,30 ,31 including increased relative abundance of Staphylococcae at baseline,31 changes in Staphylococcus species,31 and the emergence of gram-negative bacteria30 following antibiotic exposure. Two smaller studies examining oropharyngeal swabs from infants29 and children10 with CF suggested that upper airway microbial diversity increased with age. Other studies, however, including those involving this CF infant cohort, have shown that oropharyngeal microbiology does not accurately predict that of the lower airway,17 ,26 and our finding of decreasing diversity in the latter environment is therefore not surprising.
Dominance of genera of known CF pathogens was associated with reduced microbial diversity and increased pulmonary inflammation. In this cohort, bacterial density ≥105 cfu/mL was associated with greater airway inflammation.17 A recent study of otherwise healthy children with pneumonia found reduced upper airway microbial diversity,36 whilst the lower airway microbiota of adults with end-stage CF lung disease is dominated by a relatively homogenous population of typical CF pathogens.7 ,13 In cross-sectional studies of adults, increased microbial diversity is associated with clinical stability and improved pulmonary function,12 ,37 although microbial diversity was also maintained in tissue samples obtained at lobectomy from a 3 year old with severe, localised CF lung disease.38 While it was not possible to exclude the potentially confounding effect of cumulative antibiotic use, our study of young children, whose lifetime antibiotic exposure is likely considerably less than their adult counterparts, suggests that microbial diversity decreases when known pathogens emerge.
The detection of recognised CF pathogens in early childhood using conventional microbiological techniques is associated with greater airway inflammation, earlier onset of structural lung disease, and poorer pulmonary function outcomes.1–3 Using molecular techniques to confirm the former, our results clearly demonstrate that dominance of pathogen genera in 16S rRNA gene analysis is associated with greater inflammation in the lung. The impact of this finding on structural lung disease in early childhood could not be tested, as CT of the chest was not routinely performed in this patient population two decades ago. While we did not demonstrate an association between the lower airway microbiota and pulmonary function at age 6 years, normal spirometry results do not exclude bronchiectasis, which occurs in approximately 80% of early school-aged children with CF, including those with normal FEV1 values.39 Whilst this study was not designed to address causality, it is possible that longer-term follow-up will reveal a relationship between decreased diversity and abnormal pulmonary function later in life.
β diversity was of similar magnitude in longitudinally collected samples from individual subjects as it was in samples from different subjects, highlighting the extent to which the lower airway microbiota changes over time, particularly in the presence of dominant CF pathogens, where exploration of the microbiota may explain increased lower airway inflammation and clinical deterioration in the absence of conventional microbiological findings. In keeping with observations made in adult patients at various stages of an exacerbation cycle, microbial diversity may recover, either returning to baseline or developing a new microbial community structure. The long-term impact of these perturbations is unknown, but finding greater lower airway inflammation in the presence of dominant CF pathogen genera suggests a likely negative impact on structural lung disease and pulmonary function. It is unclear whether regained bacterial diversity alters the trajectory of CF lung disease in young children. Further insights can be achieved by prospective comparisons with BAL samples from age-matched controls and from analysing changes in the lower airway microbiota at frequent, more regular intervals and for a longer time period.
In conclusion, the lower airway microbiota in infants and children with CF is diverse, but bacterial diversity declines with increasing age. When present, CF pathogen genera can become dominant and are associated with greater inflammation, but individuals can recover diversity or return to their baseline microbiota. Further prospective longitudinal studies are required to understand the factors that change the lower airway microbiota over time, and to assess the impact of these changes on clinical outcome.
Acknowledgments
The authors wish to thank Liam Welsh and Suzanna Vidmar for their assistance on this project.
References
Footnotes
Contributors KBF performed the literature search, was involved in data collection and analysis, wrote the first draft of the manuscript and was involved in preparing the figures. DSA, RC and KG were authors of the previous studies of this birth cohort and were responsible for the initial study design, recruitment, data collection and analysis. SCR was responsible for the design of this study. TWF, GAS and KMW performed the 16S rRNA gene sequencing of the BAL samples. SMT analysed the 16S rRNA gene data and prepared the figures. KBF, DSA, KG, TWF, GAS, KMW, SMT and SCR were all involved in data interpretation. All authors contributed to the editing of subsequent manuscript drafts.
Funding 16S rRNA gene sequencing was funded by grants from the MCRI ‘65 km for CF’ and the RCH CF Research Trust (CFRT). KBF was supported by the Thoracic Society of Australia and New Zealand/Vertex CF Paediatric Clinical Fellowship, the RCH CFRT, the Australian CFRT Postgraduate Studentship and an Australian Government Research Training Program Scholarship. TWF, SCR GAS and KMW were supported by the National Institutes of Health (NIH) grant, HL116211 and National Health and Medical Research Council award, NHMRC1043768.
Competing interests None declared.
Ethics approval Royal Children's Hospital, Melbourne, Human Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.