Article Text

Abstract
Background Combination therapy with an inhaled corticosteroid (ICS) and long-acting β2 agonist (LABA) is recommended for patients with asthma symptomatic on ICS alone. However, there is ongoing debate regarding the risk-benefit ratio of using LABA in asthma.
Objective To evaluate the effect of the addition of a novel LABA, vilanterol (VI), to a once-daily ICS, fluticasone furoate (FF), on the risk of severe asthma exacerbations in patients with uncontrolled asthma.
Methods This randomised double-blind comparative study of variable duration (≥24–78 weeks) was designed to finish after 330 events (each patient's first on-treatment severe asthma exacerbation). 2019 patients with asthma aged ≥12 years with ≥1 recorded exacerbation within 1 year were randomised and received FF/VI 100/25 μg or FF 100 μg, administered once daily in the evening. The primary endpoint was time to first severe exacerbation; secondary endpoints were rate of severe asthma exacerbations per patient per year and change in trough evening forced expiratory volume in 1 s (FEV1) from baseline.
Results Compared with FF, FF/VI delayed the time to first severe exacerbation (HR 0.795, 95% CI 0.642 to 0.985) and reduced the annualised rate of severe exacerbations (rate reduction 25%, 95% CI 5% to 40%). Significantly greater improvements in trough FEV1 (p<0.001) were observed with FF/VI than with FF at weeks 12, 36, 52 and at endpoint. Both treatments were well tolerated with similar rates of treatment-related adverse events and on-treatment serious adverse events.
Conclusions Once-daily FF/VI reduced the risk of severe asthma exacerbations and improved lung function compared with FF alone, with good tolerability and safety profile in adolescents and adults with asthma currently receiving ICS.
ClinicalTrials.gov No NCT01086384
- Asthma
- Asthma Pharmacology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Key messages
What is the key question?
-
Does the new once-daily ICS/LABA combination FF/VI reduce the risk of severe asthma exacerbations compared with ICS alone?
What is the bottom line?
-
This randomised double-blind comparative study in patients with asthma uncontrolled on ICS confirms that the combination of FF 100 μg plus VI 25 μg reduces the risk of severe exacerbations compared with FF 100 μg alone.
Why read on?
-
This study, which employed a unique event-driven design to ensure sufficient power to study exacerbation frequency and in which most patients received treatments for >1 year, confirmed that FF/VI with its once-daily dosing regimen is more effective in reducing exacerbation risk in uncontrolled asthma than FF alone.
Introduction
The addition of a long-acting β2 agonist (LABA) to an inhaled corticosteroid (ICS) is recommended for patients whose asthma is inadequately controlled on medium-dose ICS.1 This has been shown to improve both pulmonary function and asthma control and to reduce the risk of exacerbations,2 ,3 and is the most effective and preferred option for patients requiring a step-up in asthma controller therapy, at least in adolescents and adults. However, although almost all studies have confirmed that the addition of a LABA reduces asthma exacerbations, controversy has arisen around data from a large-scale safety study4 and meta-analyses5 ,6 suggesting that more severe and life-threatening asthma events and even deaths may be increased in patients receiving LABAs. A possible explanation for this apparent increase in risk is the failure of patients being treated with LABA to receive concurrent ICS treatment.4 ,7 Indeed, this has been supported by meta-analyses comparing ICS/LABA administered as concurrent treatment (rather than from separate inhalers) with ICS alone.6 ,8 Nonetheless, this concern has resulted in reassessment of the benefits and costs of combined treatment with LABA and ICS, whether as mono-components or in combination inhalers, and for studies to examine more closely rare severe life-threatening asthma exacerbations.9 Thus, large prospective safety trials of currently marketed LABAs are underway internationally.
A new ICS/LABA combination currently being evaluated for use in asthma and chronic obstructive pulmonary disease contains fluticasone furoate (FF) and vilanterol (VI). FF is a novel ICS structurally distinct from fluticasone propionate (FP), with an ester derived from 2-furoic acid at the C-17α position that replaces the simpler propionate ester,10 conferring both greater affinity for the GC receptor and longer retention in respiratory tissues than FP.11 Once-daily FF has been shown to be effective in phase IIb trials,12–14 and the FF dose used in this study was selected on the basis of data from these studies. VI is a once-daily inhaled LABA shown to produce prolonged bronchodilation for at least 24 h. Previous clinical studies of FF/VI delivered from a single inhaler, conducted in patients with chronic obstructive pulmonary disease and including higher dosages of FF than in the present study, have shown this combination to have an acceptable safety profile.15 ,16
We report here the results of a large study, with exposure of >1 year in most patients, examining the benefits and risks of FF 100 μg and VI 25 μg administered once daily in combination in patients with asthma uncontrolled on ICS or ICS/LABA and who were consequently at increased risk of asthma exacerbations. The primary endpoint was time to first severe asthma exacerbations, as defined by the European Respiratory Society/American Thoracic Society (ERS/ATS) Task Force.17 Safety endpoints included asthma events leading to hospitalisation and intubation. Preliminary results have been presented in abstract form.18
Methods
Patients
Patients aged ≥12 years were eligible if they had a history of asthma as defined by the National Institutes for Health19 for ≥1 year prior to screening, were using ICS at a dose of ≥200 μg/day FP or equivalent or ICS/LABA at a dose of 200/100–500/100 μg FP/salmeterol or equivalent for ≥12 weeks prior to screening and at a stable dose for 4 weeks prior to screening and throughout the run-in period, and had ≥1 asthma exacerbation requiring systemic corticosteroids and/or hospital or emergency room visit in the previous year.
Eligible patients had a best prebronchodilator forced expiratory volume in 1 s (FEV1) of 50–90% predicted normal at screening, and could demonstrate ≥12% and ≥200 mL reversibility with inhaled salbutamol/albuterol. Patients’ ICS therapy was discontinued at randomisation and replaced with study medication. At randomisation, patients were required to have a recorded use of albuterol/salbutamol and/or asthma symptoms on ≥3 of the last 7 consecutive days on their daily diary.
Study design and treatments
This phase III randomised, multicentre, double-blind, parallel-group study (HZA106837; ClinicalTrials.gov registration number NCT01086384) was conducted at 167 centres in 11 countries between 22 February 2010 and 15 September 2011. After a 2-week run-in period during which baseline safety evaluations and measures of asthma status were conducted, patients were randomised (1:1) to one of two treatments administered via the ELLIPTA dry powder inhaler (GlaxoSmithKline). FF/VI 100/25 μg (representing an emitted dose from the dry powder inhaler of 92 μg for FF and 22 μg for VI) or FF 100 μg were administered once daily in the evening for a required minimum of 24 weeks and up to 78 weeks. Patients replaced their current short-acting bronchodilator and used albuterol/salbutamol as-needed for symptoms.
Patients were randomised using an automated interactive telephone-based system (RAMOS; GlaxoSmithKline, UK) in accordance with a computer-generated schedule (RandAll; GlaxoSmithKline, UK). Following randomisation, patients were not permitted to use ICS other than study medication (see online supplementary appendix for a full list of permitted and prohibited medications).
An event-driven design was employed, meaning that the study was planned to finish after 330 ‘events’ had occurred. An event was defined as a patient's first severe asthma exacerbation in the study. A severe asthma exacerbation was defined using the ERS/ATS Task Force recommendation as a deterioration of asthma requiring the use of systemic corticosteroids for at least 3 days, or inpatient hospitalisation, or emergency department visit due to asthma requiring systemic corticosteroids.17 A blinded independent adjudication committee ensured that all severe asthma exacerbations were captured as defined in the protocol. Only events deemed by the adjudication committee to be severe asthma exacerbations were used in the endpoint analysis.
One interim analysis was performed to evaluate the primary endpoint (ie, time to first severe asthma exacerbation) and to identify any potential treatment harm by reviewing the most frequent on-treatment adverse events and asthma-related mortality/morbidity (see online supplementary appendix).
Outcome measurements
The primary endpoint was time to first severe asthma exacerbation. Secondary efficacy endpoints were rate of severe asthma exacerbations per patient per year and change from baseline at week 36 in evening trough FEV1. Other endpoints are listed in the online supplementary appendix.
Safety
Safety endpoints relating to severe asthma exacerbations included the number of hospitalisations, emergency department/urgent care visits, unscheduled healthcare provider visits and intubations for an asthma event. General safety and tolerability endpoints including vital signs were monitored (see online supplementary appendix).
Patients were withdrawn from the study if they experienced three on-treatment severe asthma exacerbations in any 6-month period or four throughout the treatment period.
Statistical analysis
Three hundred and thirty events were required to provide 90% power to detect a 30% reduction in risk (HR 0.70) of severe asthma exacerbation at a two-sided significance level of 0.05. The total sample size of 2000 (1000 per treatment arm) was based on assumptions of 10% loss to follow-up, 20% of patients in the FF arm having ≥1 severe asthma exacerbations per year and a recruitment pattern as specified in the protocol.
The primary efficacy endpoint was analysed by Cox proportional hazards analysis (FF/VI vs FF) of time to first severe asthma exacerbation, incorporating terms for baseline FEV1, sex, age and region. A Cox proportional hazards analysis was performed to examine treatment interactions with these covariates. Statistical methods for the secondary and other efficacy endpoints and sensitivity analyses including the interim analysis of the primary efficacy endpoint are described in the online supplementary appendix. All efficacy and safety analyses were carried out in the intent-to-treat (ITT) population other than those specified as being carried out in the per protocol (PP) population (see online supplementary appendix). The decision to exclude a patient or some of a patient's data from the PP population was made prior to breaking the blinding.
Results
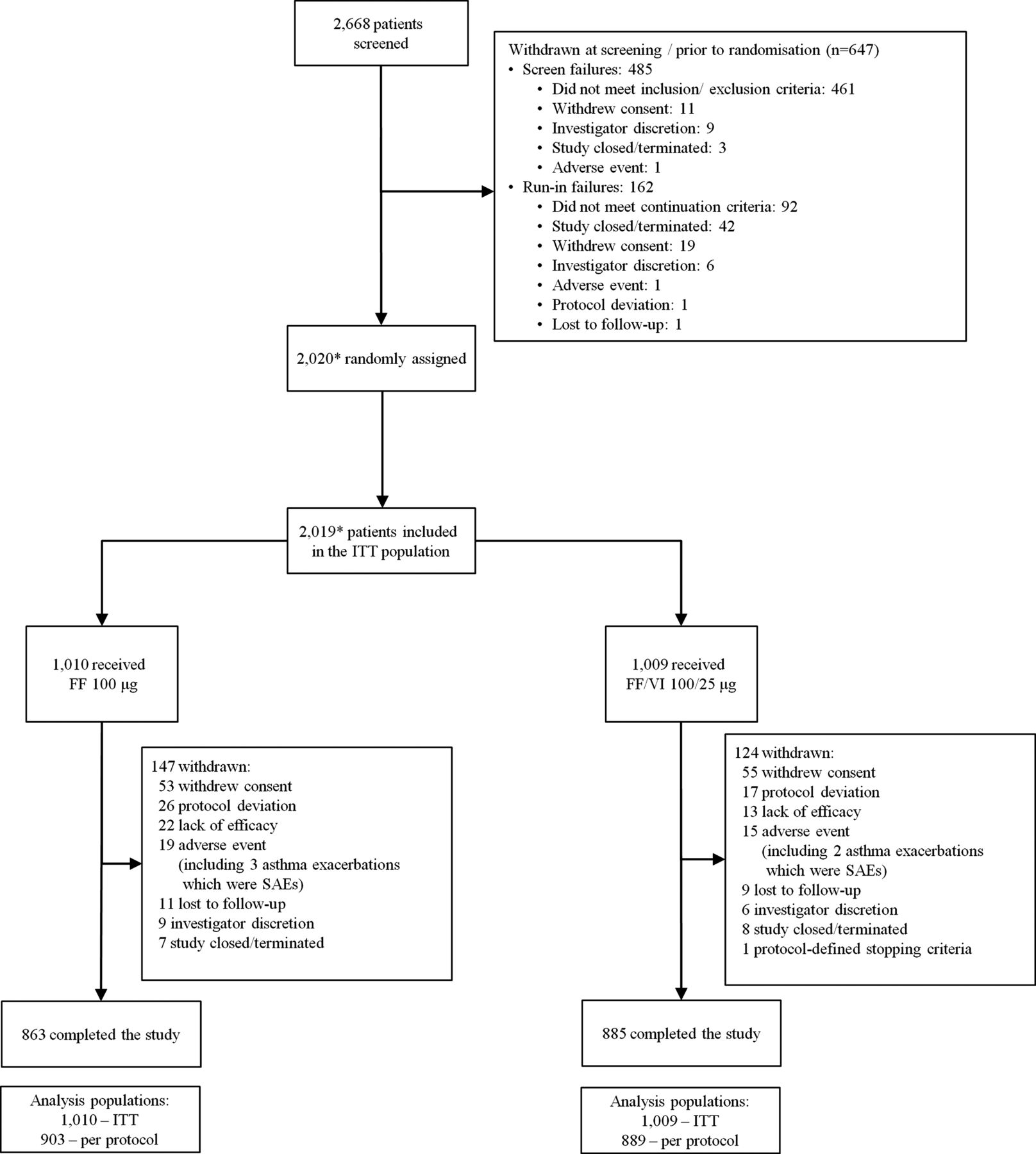
Of 2668 patients screened, 2020 were randomised and 2019 comprised the ITT population (figure 1). The PP population consisted of 1792 patients (89% of the ITT population): 903 received FF 100 μg and 889 received FF/VI 100/25 μg. Demographics and baseline characteristics were generally similar across the treatment groups in the ITT (table 1 and online supplementary table E1) and PP populations. All patients were taking ICS at baseline and approximately 60% of patients were also receiving a LABA either as a separate inhaler or as part of an ICS/LABA combination product. Adolescents (age 12–17 years) comprised 14% of the ITT population.
Patient demographics and baseline characteristics, intent-to-treat population

Patient disposition and reasons for withdrawal post-screening. *One patient was not randomised but received FF 100 μg in error and one patient was randomised but did not receive treatment; these patients are not included in the ITT population. The patient who received FF 100 μg in error received 5 days of treatment and then was withdrawn. No safety issues were identified during this treatment period; FF, fluticasone furoate; ITT, intent-to-treat; SAE, severe adverse event; VI, vilanterol.
Due to the event-driven study design, the duration of exposure to study treatment was variable. A minimum duration of active treatment of 24 weeks was planned; only patients who were either withdrawn by the investigator or withdrew voluntarily had <24 weeks of treatment. All patients who completed the planned double-blind treatment period were treated for ≥24 weeks and no patients were treated for longer than 78 weeks. The mean duration of treatment exposure (52.0–52.7 weeks) and the proportion of patients who received treatment for ≥52 weeks (56–58%) were similar between the groups (see online supplementary figure E1).
Primary efficacy and related endpoints
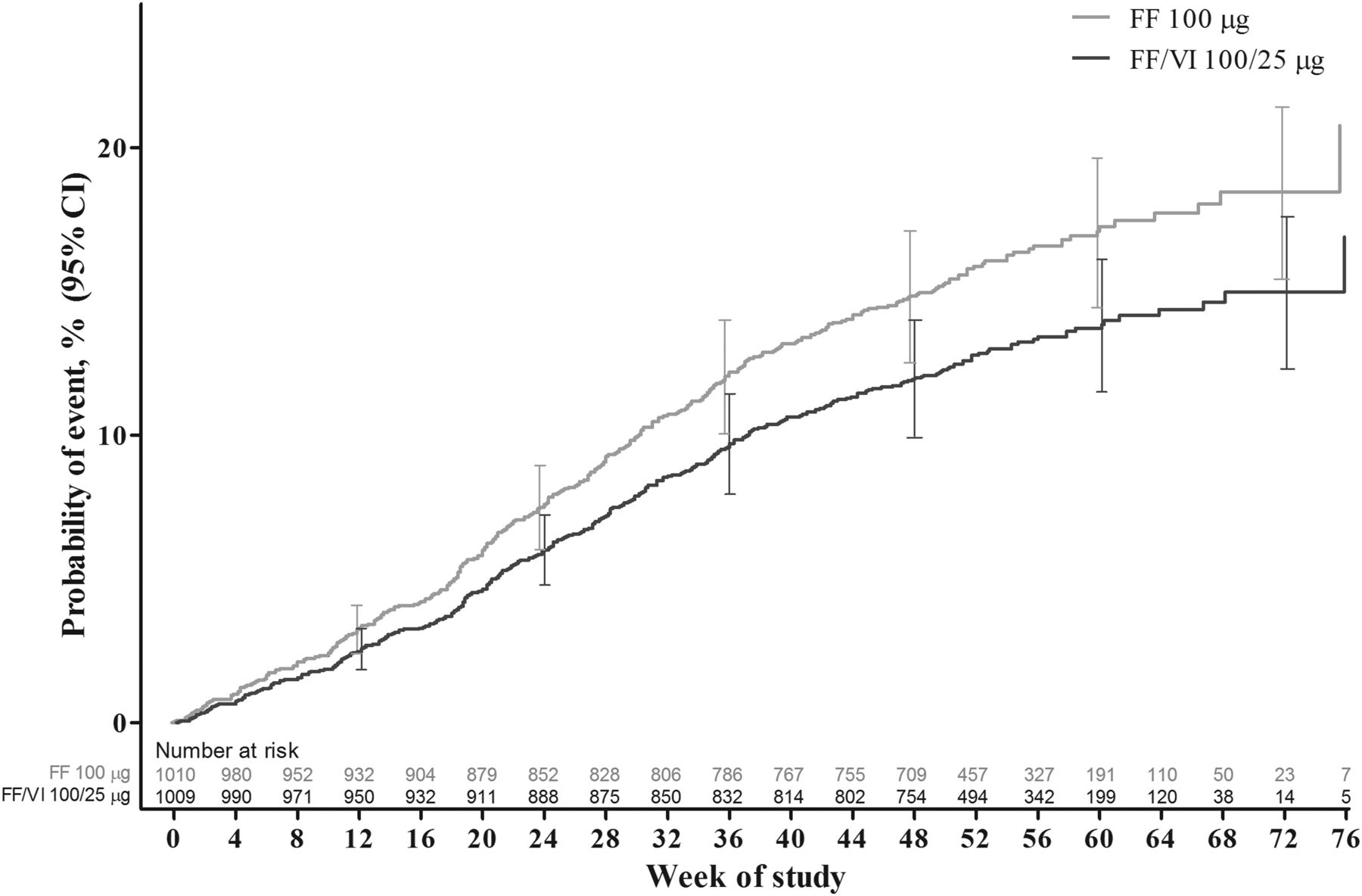
FF/VI significantly delayed the time to the first severe asthma exacerbation relative to FF (table 2, figure 2). The adjusted probability of experiencing a severe asthma exacerbation by 52 weeks was 15.9% (95% CI 13.5% to 18.2%) in the FF 100 μg group and 12.8% (95% CI 10.7% to 14.9%) in the FF/VI 100/25 μg group. The HR for FF/VI 100/25 μg vs FF 100 μg was 0.795 (95% CI 0.642 to 0.985, p=0.036, adjusted for the interim analysis), representing a 20% risk reduction. Analysis of the PP population provided similar results: HR (FF/VI vs FF) was 0.722 (95% CI 0.548 to 0.950), representing a 28% risk reduction (p=0.020). A total of 340 patients experienced ≥1 severe exacerbations; >99% of the severe asthma exacerbations entered into the case report form were confirmed by the adjudication committee. The outcomes of subgroup analyses of treatment interactions with baseline FEV1, age, sex and region showed a statistically significant (p<0.10) interaction between baseline FEV1 and treatment (see online supplementary figure E2). Interactions between treatment and the remaining factors (age, sex and region) were not statistically significant.
Cox proportional hazards analysis of time to first severe asthma exacerbation, intent-to-treat population
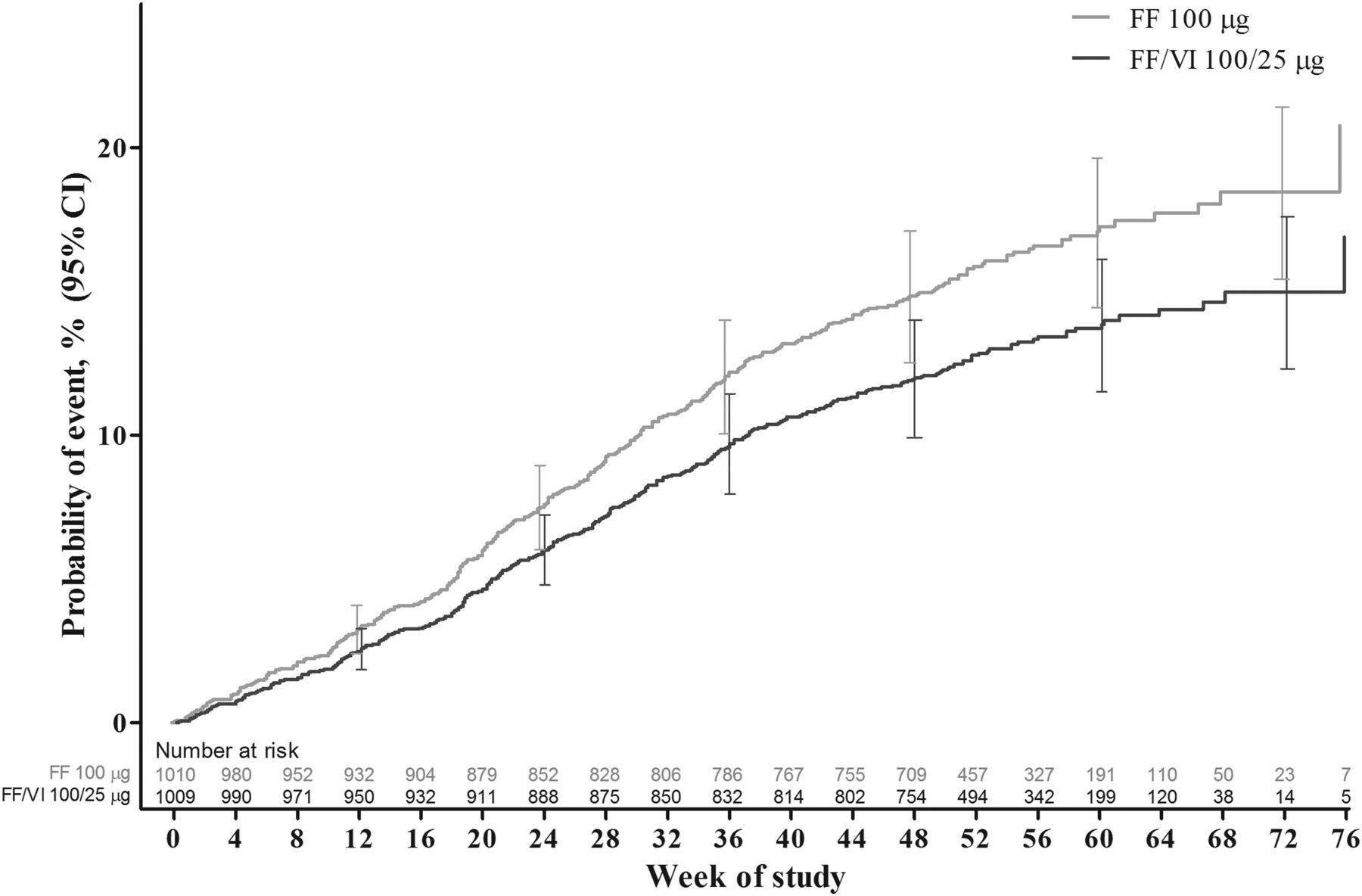
Cox proportional hazards model cumulative incidence curve for time to first severe asthma exacerbation, intent-to-treat population. FF, fluticasone furoate; VI, vilanterol.
The possibility of informative censoring was explored. The Kaplan–Meier curves for time to first severe asthma exacerbation or withdrawal due to lack of efficacy, investigator discretion or withdrawal of consent between the two treatment groups did not differ from those of the primary analysis (see online supplementary figure E3).
Secondary and other efficacy endpoints
The rate of severe asthma exacerbations per patient per year was significantly lower in the FF/VI 100/25 μg group than in the FF 100 μg group (0.14 vs 0.19), a reduction in rate of 25% (95% CI 5% to 40%; p=0.014). The number of patients experiencing ≥1 on-treatment severe asthma exacerbation was also lower in the FF/VI group: 186 patients (18%) with FF (271 exacerbations in total) versus 154 patients (15%) with FF/VI (200 exacerbations in total). The mean duration of a severe asthma exacerbation was 11 days in both groups. Online supplementary figure E4 shows the number and duration of severe asthma exacerbations experienced by individual patients during the study.
Trough FEV1 increased over the treatment period in both the FF and FF/VI treatment groups (figure 3 and online supplementary figure E5). FF/VI demonstrated statistically significant improvements over FF in trough FEV1, with adjusted mean changes of 83–95 mL (p<0.001).
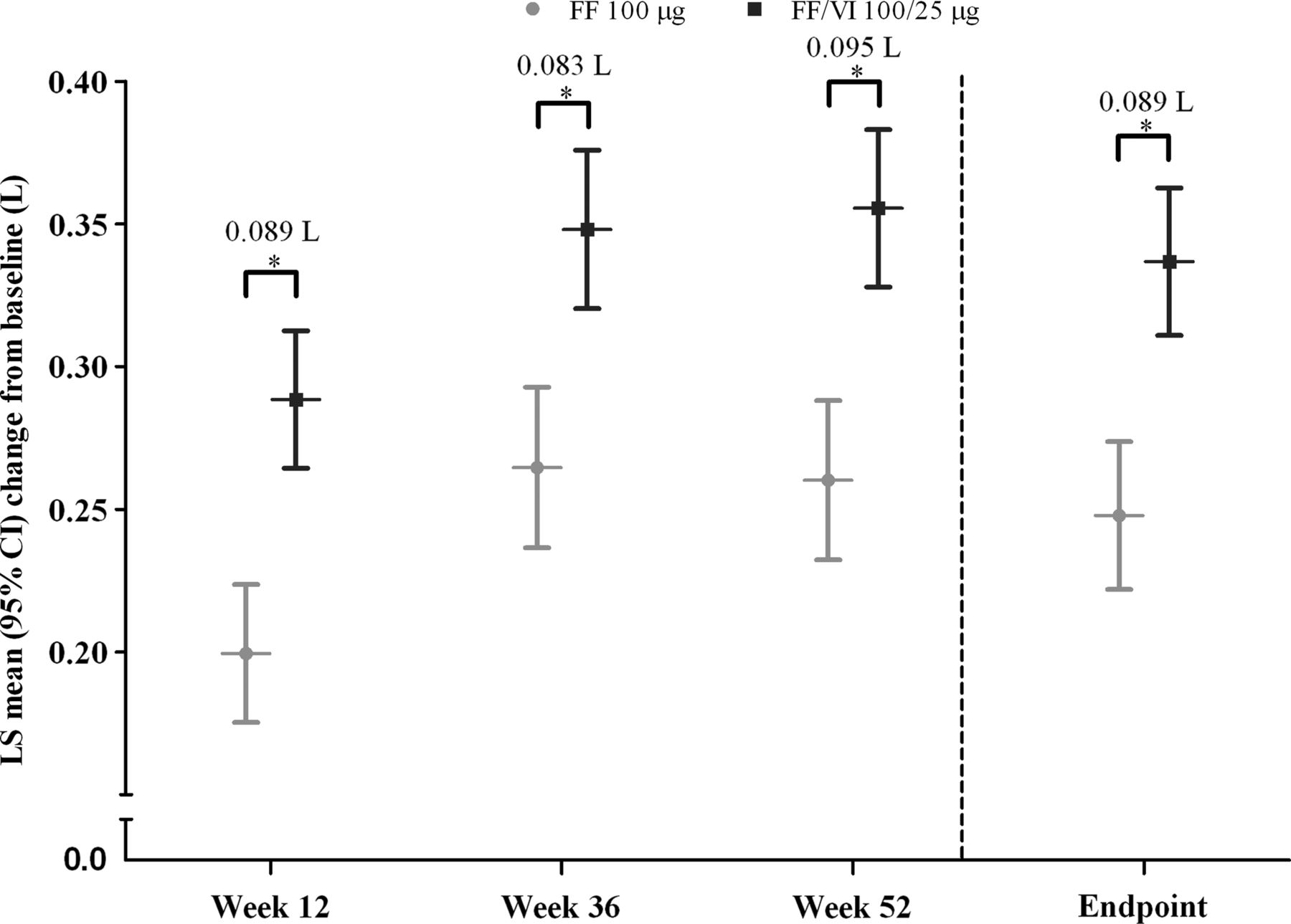
Adjusted mean changes from baseline in trough forced expiratory volume in 1 s (L), intent-to-treat population. *Treatment differences p<0.001. FF, fluticasone furoate; LS, least squares; VI, vilanterol.
Self-reported rescue albuterol/salbutamol use increased over the 14 days preceding an exacerbation (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean self-reported daily rescue use (albuterol/salbutamol) 14 days before and after the onset of severe asthma exacerbation for patients who experienced ≥1 or 0 severe asthma exacerbations, intent-to-treat population. *Date of onset was determined by the investigator and recorded in their clinical notes. For patients who experienced ≥1 severe asthma exacerbation, all severe asthma exacerbations are included in the figure. Rescue use in patients who did not exacerbate during the study was calculated for the same duration (number of days) as each exacerbation event in exacerbating patients. For this purpose, the surrogate Day 0 for non-exacerbators was the median study day of onset for all exacerbations. FF, fluticasone furoate; VI, vilanterol.
Adjusted mean changes from the baseline Asthma Control Questionnaire (ACQ7) are shown in online supplementary figure E6. Significantly greater improvements in the ACQ7 score were observed in patients receiving FF/VI compared with FF at all time points (p<0.001; week 12, week 36 and endpoint). The proportion of patients with well-controlled asthma (ACQ7 score ≤0.75) at baseline was similar in the two treatment groups (both 2%). ORs for FF/VI versus FF at week 12 (1.49, 95% CI 1.20 to 1.84), week 36 (1.49, 95% CI 1.21 to 1.83) and at endpoint (1.50, 95% CI 1.23 to 1.82) indicated that patients in the FF/VI group were significantly more likely to be well controlled than those in the FF group (all p<0.001). At endpoint, more patients in the FF/VI group than the FF group were well controlled (44% vs 36%).
Safety assessment
Similar proportions of patients experienced severe asthma exacerbations leading to hospitalisation with FF (n=9, <1%) and FF/VI (n=8, <1%). In the FF group, 26 (3%) patients visited an emergency department or urgent care clinic due to a severe asthma exacerbation and 142 (14%) made unscheduled visits to a healthcare provider. These frequencies were 22 (2%) and 119 (12%), respectively, for FF/VI. No patients were intubated due to a severe asthma exacerbation.
FF and FF/VI had similar overall safety profiles (table 3). There were 29 (3%) on-treatment serious adverse events (SAEs) in the FF group (seven considered asthma-related by the adjudication committee) and 41 (4%) in the FF/VI group (10 considered asthma-related). Four on-treatment and post-treatment SAEs were deemed treatment-related, three (pleurisy, asthma, non-cardiac chest pain) in the FF group and one (tachyarrhythmia) in the FF/VI group. Three fatalities occurred: two (pneumonia, metastatic lung cancer (post-treatment)) in the FF group and one (automobile accident as passenger) in the FF/VI group; none were deemed to be treatment-related or asthma-related by the investigators or adjudication committee.
Most frequent (≥5%) on-treatment AEs, intent-to-treat population
Small but statistically significant treatment differences in change from baseline in diastolic blood pressure were observed at week 44 (–0.8 mm Hg, p=0.022) and week 76/end of study (–0.7 mm Hg, p=0.032); however, these were not considered to be clinically important. No statistically significant treatment differences in systolic blood pressure or pulse rate were observed.
Discussion
This study showed that, in patients with asthma uncontrolled on ICS, FF/VI 100/25 μg once daily administered for up to 78 weeks reduced the risk of experiencing a severe asthma exacerbation by 20% and reduced the rate of severe exacerbations per patient per year by 25% compared with FF 100 μg alone. FF/VI also significantly improved trough FEV1 and the number of patients achieving well-controlled asthma. FF/VI had a good safety profile without evidence of life-threatening asthma events. Reducing asthma exacerbations is considered in asthma guidelines to be the most important endpoint of future risk, and is of considerable benefit to patients because of their impact on quality of life and the high healthcare costs associated with their management. Although increasing the dose of ICS has been shown to be highly effective at reducing exacerbation risk, this approach needs to be considered in conjunction with the potential side effects of long-term use of higher doses of ICS.20
The results with FF/VI are consistent with those of many studies, confirming that the addition of a LABA to a medium dose of ICS improves lung function and asthma control and reduces the risk of severe asthma exacerbations including hospitalisations.1 ,21–23 However, our study is the first to demonstrate this improvement with once-daily dosing. We did not compare the efficacy of FF/VI treatment with doubling the dose of FF in this cohort of patients with uncontrolled asthma, but results from studies with that design have provided clear evidence of greater clinical benefit of combination therapy compared with doubling the dose of ICS.24–26 Unlike FP and beclametasone dipropionate, for which three doses are available for use in adults, two doses of FF will be available for use in adults/adolescents. FF 100 μg is suitable for use in patients who require FP 100–250 μg twice daily and FF 200 μg is suitable for patients who require FP 500 μg twice daily or equivalent. Therefore, in this study, some patients will have been randomised to a similar ICS dose to baseline and some may have received a reduced ICS dose.
In this study the results of secondary endpoints, including annualised rate of severe exacerbations, further support the primary endpoint. The improvements in trough FEV1 and reductions in ACQ7 scores confirm superior current asthma control with the FF/VI combination. In addition, the frequency of severe asthma exacerbations leading to hospitalisation, emergency room visit or unscheduled healthcare provider visit was similar for both FF and FF/VI. Thus, there was no indication of an increased risk of such severe events with the addition of the LABA, a finding that is consistent with several meta-analyses of studies comparing combined ICS/LABA with ICS alone.6 ,7 ,23
Interpreting the clinical significance of exacerbation reductions in clinical trials is easiest when the comparator is usual treatment. In this study two new treatments were compared: a new once-daily moderate dose of FF and FF combined with VI. The resultant annualised rate of severe asthma exacerbations was low in both treatment groups, 0.19 for FF (corresponding to approximately one exacerbation every 5 years per patient) and 0.14 for FF/VI (one every 7 years), despite the fact that all patients were required to have had a severe asthma exacerbation during the 1-year period prior to randomisation. The exacerbation rate observed in the FF group compares favourably with those observed in previous studies of patients uncontrolled on medium-dose ICS, in which annualised rates ranging from 0.31 to 0.35 were reported for patients receiving budesonide alone.25 ,27 ,28 In two recent studies of the effect of adding salmeterol to FP on asthma exacerbation rates,29 ,30 rates of 0.27–0.30 exacerbations/patient/year were reported for patients using FP monotherapy. Thus, the 25% decrease seen in this study represents a clinically useful improvement from an already very low base rate.
Regarding safety, FF and FF/VI were well tolerated and the incidence of treatment-related adverse events and all SAEs was low and similar across treatment groups. No clinically relevant treatment differences in vital signs or liver function parameters were observed. In view of the concerns regarding a possible link between LABA use and asthma-related hospitalisations and fatal events,4 no association was observed in this study of >2000 patients with a mean exposure of ≥12 months, although we recognise that this cannot be viewed as conclusive evidence.
The current ERS/ATS Task Force definition of severe asthma exacerbations17 was used, and an adjudication committee provided a blinded review to ensure that all severe asthma exacerbations were identified and included in the primary measure. In addition, the monitoring of rescue use preceding and during the time of each severe asthma exacerbation confirmed that exacerbations were preceded by a period of loss of asthma control, supporting the accuracy and appropriateness of recording of asthma exacerbations.31
Strengths of this study include its innovative design in which the duration was not predetermined. Instead, the study was terminated when a specified number of acute exacerbations had occurred. In addition, most patients remained in the study and received treatment for 52 weeks or more, thus permitting a reliable determination of the annualised rate of severe asthma exacerbations. In contrast, in studies of fixed duration, although nominally of 12 months’ duration, mean exposures to study drugs are reduced owing to premature withdrawals, particularly for patients who experience exacerbations. However, the event-driven design makes it difficult to compare the results of the present study with those of fixed duration. In our study, informative censoring did not occur differentially between the two treatment arms.
The dose of FF/VI (100/25 μg) used in this study was selected on the basis of data from earlier phase dose-ranging studies.12 ,13 ,32 FF 100 μg was used as the comparator, ensuring that all patients were treated with at least a mid-strength ICS throughout the study. This study was not designed to compare the effect of adding a LABA with that of increasing the dose of ICS. The study did not examine the effect of time of dosing. Finally, the study population exhibited marked bronchodilator responsivenes to a β2 agonist (salbutamol) at the screening visit (approximately 500 mL and 22% of baseline), although only 200 mL and 12% was required for inclusion.
In summary, this study confirms that the combination of FF/VI 100/25 μg administered once daily in the evening to adolescents and adults with moderate asthma significantly reduced the risk of severe asthma exacerbations, improved lung function and led to asthma control in a larger proportion of patients than FF 100 μg. Both treatments were well tolerated with similar safety profiles and a low incidence of treatment-related AEs and SAEs, and no increased risk of serious asthma-related events was seen with the addition of VI.
Acknowledgments
We thank all patients and investigators involved in the study, and the members of the adjudication committee and Independent Data Monitoring Committee. Editorial support (in the form of development of a draft outline in consultation with the authors, development of a manuscript first draft in consultation with the authors, editorial suggestions to draft versions of this paper, assembling tables and figures, collating author comments, copyediting, fact checking, referencing and graphic services) was provided by Ian Grieve, PhD at Gardiner-Caldwell Communications (Macclesfield, UK) and was funded by GlaxoSmithKline.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors All authors, including the independent steering committee (EDB, PMO'B, WWB, JLö, ERB, AW) together with authors employed by the sponsor (LA, LJ, LF, JLi) had access to all of the data, were involved in every stage of the preparation of the paper and approved the final version. Employees of the sponsor (led by LF and JLi) performed the statistical analysis. The sponsor did not place any restriction on authors about the statements made in the final paper. All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors.
-
Funding This study was funded by GlaxoSmithKline (study number HZA106837; ClinicalTrials.gov identifier NCT01086384).
-
Competing interests EDB has served as a consultant to AlkAbello, Almirall, Cephalon, Hoffman la Roche, ICON and MS Consulting Group; has been on advisory boards for Almirall, AstraZeneca, Boehringer Ingelheim, Elevation Pharma, Forest, GlaxoSmithKline, Merck, Napp, Novartis and Nycomed; has received lecture fees from AlkAbello, AstraZeneca, Boehringer Ingelheim, Chiesi, GlaxoSmithKline, Novartis, Pfizer and Takeda; and his institution has received remuneration for participation in clinical trials sponsored by Actelion, Aeras, Almirall, AstraZeneca, Boehringer Ingelheim, Forest, GlaxoSmithKline, Hoffman La Roche, Merck, Novartis, Takeda and TEVA. PMO'B has served as a consultant to AstraZeneca, Almirall, Boehringer Ingelheim, GlaxoSmithKline and Merck; has served on advisory boards for AIM, Altair, Boehringer, GlaxoSmithKline, Medimmune and Merck; has received lecture fees from Chiesi; and has received research funding from Amgen, AstraZeneca, Asmacure, Genentech and Ono. WWB has served as a consultant to Amgen, AstraZeneca, Boehringer Ingelheim, Genentech, GlaxoSmithKline, MedImmune, Novartis and TEVA; has served on advisory boards for Altair, Amgen, Centocor, GlaxoSmithKline, Johnson & Johnson, Merck Sharpe and Dohme and Pfizer; has received lecture fees from Merck Sharpe and Dohme; and has received research funding from AstraZeneca, Ception, GlaxoSmithKline, MedImmune and Novartis. JLö has served as a consultant to and received lecture fees from AstraZeneca, GlaxoSmithKline, Merck Sharpe and Dohme, Novartis, and UCB Pharma; has been partly covered by some of these companies to attend previous scientific meetings including the ERS and the AAAAI; has provided expert testimony for Barr Pharmaceuticals; and has participated in clinical research studies sponsored by AstraZeneca, GlaxoSmithKline, Merck Sharpe and Dohme and Novartis. ERB has served as a consultant to AstraZeneca, Boehringer Ingelheim, Cephalon, Forest, Genentech, GlaxoSmithKline, MedImmune, Novartis, Pfizer and Sanofi-Aventis; has received reimbursement for travel from GlaxoSmithKline; and has performed clinical trials for AstraZeneca, Boehringer Ingelheim, Centocor, Genentech, GlaxoSmithKline, Johnson & Johnson, Merck, Novartis and Pfizer which have been administered by his employer Wake Forest University School of Medicine. AW has served as a consultant to Almirall, AstraZeneca, Chiesi, Cytos, GlaxoSmithKline, Merck Sharpe and Dohme and Novartis; and has received lecture fees and research grants from Chiesi and GlaxoSmithKline. LA, LJ, LF and JLi are employees of and hold stock in GlaxoSmithKline.
-
Patient consent Obtained.
-
Ethics approval The study was approved by local ethics review committees and was conducted in accordance with the Declaration of Helsinki, Good Clinical Practice guidelines and all applicable regulatory requirements.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Airwaves