Article Text

Abstract
Background Distal cell-type-specific regulatory elements may be located at very large distances from the genes that they control and are often hidden within intergenic regions or in introns of other genes. The development of methods that enable mapping of regions of open chromatin genome wide has greatly advanced the identification and characterisation of these elements.
Methods Here we use DNase I hypersensitivity mapping followed by deep sequencing (DNase-seq) to generate a map of open chromatin in primary human tracheal epithelial (HTE) cells and use bioinformatic approaches to characterise the distribution of these sites within the genome and with respect to gene promoters, intronic and intergenic regions.
Results Genes with HTE-selective open chromatin at their promoters were associated with multiple pathways of epithelial function and differentiation. The data predict novel cell-type-specific regulatory elements for genes involved in HTE cell function, such as structural proteins and ion channels, and the transcription factors that may interact with them to control gene expression. Moreover, the map of open chromatin can identify the location of potentially critical regulatory elements in genome-wide association studies (GWAS) in which the strongest association is with single nucleotide polymorphisms in non-coding regions of the genome. We demonstrate its relevance to a recent GWAS that identifies modifiers of cystic fibrosis lung disease severity.
Conclusion Since HTE cells have many functional similarities with bronchial epithelial cells and other differentiated cells in the respiratory epithelium, these data are of direct relevance to elucidating the molecular basis of normal lung function and lung disease.
- Human tracheal epithelium
- open chromatin
- cis-acting regulatory elements
- lung cancer
- non-small cell lung cancer
- airway epithelium
- cystic fibrosis
- paediatric lung disease
Statistics from Altmetric.com
- Human tracheal epithelium
- open chromatin
- cis-acting regulatory elements
- lung cancer
- non-small cell lung cancer
- airway epithelium
- cystic fibrosis
- paediatric lung disease
Key messages
What is the key question?
What are the regulatory elements that control gene expression in the airway epithelium and where in the genome are they located?
What is the bottom line?
We describe a resource that will enable rapid identification of functional genetic elements that control transcriptional networks in the airway epithelium.
Why read on?
This has immediate relevance to determining the normal differentiation of the airway epithelium and what goes wrong in airway disease.
Introduction
The epithelia that line the trachea and bronchi of the human airway have important functions in protecting the respiratory system from environmental insults and pathogenic organisms while maintaining the conduit for air to and from the alveoli. Furthermore, major lung diseases such as chronic obstructive pulmonary disease (COPD), asthma and cystic fibrosis are associated with malfunction of the airway epithelium. The epithelial layer interacts closely with other cells in the airway, such as endothelial and immune cells. This critical cell layer requires the establishment and maintenance of coordinate patterns of gene expression, which ensure structural differentiation of the epithelium, including apical/basolateral polarity, integration of the mucociliary escalator and appropriate regulation of ion transport. Though the regulatory mechanisms for many individual genes that are active in the airway epithelium were characterised by classical methods, other genes, particularly those which show tight cell-type-specific control involving cis-acting elements that lie outside the promoter or coding region, are less well understood. Until recently, the cellular machinery that achieves coordinated function of airway epithelium has been hard to evaluate since this requires global analysis of regulatory elements in relatively small numbers of differentiated primary airway epithelial cells. However, techniques developed in part by the ENCODE consortium,1 including methods to identify regions of open chromatin genome wide, such as DNase I hypersensitivity mapping followed by deep sequencing (DNase-seq)2 and formaldehyde-assisted identification of regulatory elements (FAIRE),3 have enabled these analyses. Here we describe a genome-wide map of open chromatin in primary human tracheal epithelial (HTE) cells generated by DNase-seq analysis. We use bioinformatic tools to identify tracheal epithelial-cell-selective regions of open chromatin and determine their distribution throughout the genome. We determine that the peaks of open chromatin are more evident at the promoters of genes that are highly expressed in HTE cells than at inactive gene promoters. Moreover, HTE-selective peaks of open chromatin are associated with genes involved in pathways of epithelial differentiation and function. Within these peaks, predicted binding sites for some epithelial-specific transcription factors are over-represented. Next, we illustrate the power of these data to facilitate the characterisation of regulatory elements for genes that are coordinately expressed in HTE cells, such as structural proteins and ion channels. Finally, we demonstrate the use of these data to identify the molecular basis of genome-wide association studies (GWAS) that identify non-coding regions of the genome as strong candidates for disease effectors or modifiers. These results will enhance the understanding of transcriptional networks that coordinate lung epithelial function in health and disease.
Results
Identification of DNase hypersensitive sites genome-wide
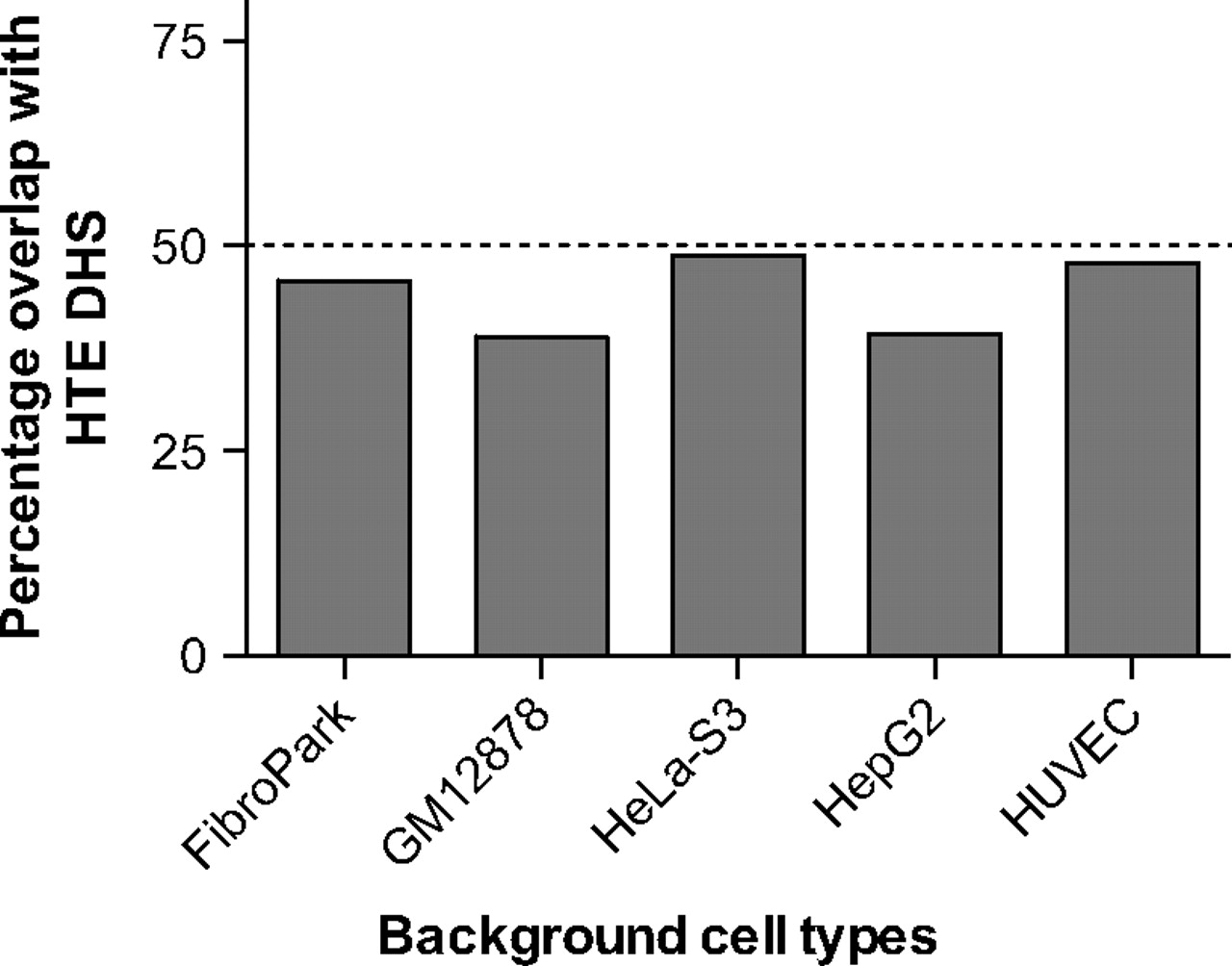
HTE cells were evaluated for regions of open chromatin genome-wide by mapping DNase I hypersensitive sites using DNase-seq. The sequence reads were then analysed with the F-seq application, a feature density estimator for high-throughput sequence tags,4 which identified 153 504 DNase hypersensitive sites (DHS) in the HTE cells. These sites represent elements in the genome where multiple sequence reads (peak signals) aligned to a common region. Since regulatory elements are often located within DHS, this map of open chromatin has the potential to identify regulatory elements for all genes that are expressed in the HTE cells. To distinguish between ubiquitous and cell-type-selective regulatory elements, we next subtracted DNase-seq data sets from five different cell types generated by the ENCODE consortium.1 These included skin fibroblasts (FibroPark), a lymphoblastoid cell line (GM12878), a cervical carcinoma cell line (HeLa-S3), a liver carcinoma cell line (HepG2), and human umbilical vein endothelial cells (HUVEC). Though two of these lines are carcinomas, open chromatin data from relevant primary cells are not available for these epithelial cell types. The genomic overlap between HTE DHS and the other five cell types is shown in figure 1 and the numbers of DHS are shown in table 1. Of the 153 504 HTE sites, 39 656 (25.8%) were found to be cell type selective, in that they did not intersect with the hypersensitive sites present in any of the five other cell types, and 31 146 (20.3%) sites were ubiquitous, in that they overlapped (at least partially) with DHS from all five of the other cell types. Further analysis of the HTE DHS across different cell types revealed 974 National Center for Biotechnology Information (NCBI) Reference Sequence (RefSeq) genes with a greater representation of DHS within their promoters and coding regions than the five background cell types combined (see online supplementary table 1). Many of these genes code for proteins that have well characterised functions in the airway epithelium and some of these are discussed further below.

Cell-type specificity of DNase I hypersensitive sites (DHS) in human tracheal epithelial (HTE) cells. The genomic overlap between HTE DHS and the other five cell types measured as the percentage of HTE DHS that overlap with each of the individual background cell types. FibroPark, skin fibroblasts; GM12878, a lymphoblastoid cell line; HeLa-S3, a cervical carcinoma cell line; HepG2, a liver carcinoma cell line; HUVEC, human umbilical vein endothelial cell.
Number of open chromatin sites in human tracheal epithelial cells and the six other cell types that were used for comparison
Distribution of DHS across the genome
Next we analysed the distribution of DHS with respect to different genomic elements, according to the following categories: promoter (2 kb of sequence 5′ to the transcriptional start site), exon 1, intron 1, other genic, 2 kb of sequence 3′ to the transcriptional stop site, and intergenic (figure 2). DHS were subdivided according to whether they were HTE selective or ubiquitous (as defined above) and a third group (genome wide), which included all sites. We observed that the ubiquitous sites occur more frequently (25.6%) in promoter regions than HTE-selective sites (2.5%) and genome-wide sites (1.2%). A similar distribution of DHS is seen in the first exon of genes and to a lesser extent in the first introns and 2 kb downstream of genes. In contrast, the HTE-selective sites are most common in other genic sites and also more common than ubiquitous sites in intergenic regions.

Human tracheal epithelial (HTE)-selective DNase I hypersensitive sites (DHS) are generally located in distal regions of genes or in intergenic sequences, rather than in promoters, where ubiquitous DHS predominate. Three categories of DHS (all DHS, HTE-selective DHS, and ubiquitous DHS) were overlapped with different genomic regions to determine their distribution with respect to genes. 2 kb up, including 2 kb 5′ to genes; other genic, all exons and introns of genes excluding the first; 2 kb down, including 2 kb 3′ to genes; intergenic, all genomic sequence more than 2 kb from genes.
Correlation of DNase-seq data with gene expression
Total RNA was extracted from the same cell cultures that were used for DNase-seq and evaluated on Nimblegen 72K HG18 60mer microarrays to characterise gene expression profiles. To look for a correlation between gene expression in HTE cells and the DNase-seq signal, gene expression values were divided into three groups: high expression (top 20%), middle expression (middle 20%) and low expression (bottom 20%). The remaining 40% of gene expression values were not included in the analysis. The DNase-seq base overlap signal, which is the number of reads that align to each base pair position of the genome, was averaged across the 1 kb before and after the transcription start site of the genes within each of these categories (figure 3). The data show that the most highly expressed genes correlate with an increased DNase-seq signal.

The intensity of DNase I hypersensitive sites (DHS) at gene promoters correlates with gene expression in human tracheal epithelial (HTE) cells. Genes for which microarray expression data were available were separated into three categories: high expression (top 20%), mid expression (middle 20%), and low expression (bottom 20%). Then, the average number of DNase-se reads was calculated at each base between 1 kb 5′ and 1 kb 3′ to the transcription start site (TSS) for each of these categories.
Pathways of epithelial structure and function revealed by HTE-selective promoter DHS
The Entrez Gene IDs for the 1061 genes that exhibited one or more HTE-selective promoter DHS (representing 1118 DHS) were compiled into a text file and analysed with DAVID (Database for Annotation, Visualisation and Integrated Discovery)5 to look for gene processes that might be over-represented compared with all human genes. The top 10 most statistically significant DAVID ontologies/pathways are shown in table 2 and the entire list (p<0.1) is presented in online supplementary table 2. Five of the top 10 DAVID results are directly related to epithelial function, including epithelial cell differentiation (GO:0030855), epithelium development (GO:0060429), ectoderm development (GO:0007398), epidermis development (GO:0008544), and epidermal cell differentiation (GO:0009913). Moreover, three additional pathways are relevant to the function of polarised cells in the epithelial sheet lining the airway, including cell junction (GO:0030054), apicolateral plasma membrane (GO:0016327) and apical junction complex (GO:0043296). This analysis thus validates the use of DNase-seq to identify cell-type-specific regulatory elements that are associated with open chromatin in HTE cells.
Top 10 statistically over-represented processes from Database for Annotation, Visualisation and Integrated Discovery (DAVID) analysis when comparing a list of genes with human tracheal epithelial (HTE)-selective DNase I hypersensitive sites in their promoter to all human genes
HTE-selective COREs overlapping promoter regions identify genes with multiple functions in airway epithelial cells
Next we took a global view of HTE-selective DHS and observed that these tended to be clustered, rather than being randomly distributed throughout the genome, as has previously been observed for islet-cell-selective regions of open chromatin, defined by FAIRE.6 Following their definition of a CORE (cluster of open regulatory elements, a cluster of three or more discrete regions of open chromatin separated by <20 kb) we identified 3608 HTE-selective COREs. We next evaluated the 998 HTE-selective COREs that overlapped with the promoter regions (2 kb upstream of transcriptional start sites) of 1328 genes (see online supplementary table 3). An additional data set (of 1975 COREs) was generated for this analysis that subtracted the open chromatin map for normal human epidermal keratinocytes (NHEK) since these too are differentiated primary epithelial cells and we wanted to distinguish between genes involved in epithelial function generally and HTE cells specifically (the overlap between HTE and NHEK is greater than that of the other cell types examined in DHS; see online supplementary figure 1). Microarray data for RNA abundance derived from the same HTE samples that were used to map open chromatin were incorporated into this analysis and we evaluated in detail the top 500 expressed genes associated with HTE-selective COREs overlapping their promoters. This analysis is clearly biased towards identification of HTE-selective COREs that are associated with transcriptional activation and not repression. Multiple genes were identified that were relevant to airway epithelial function, including structural proteins, ion channel components and transcription factors. Among structural proteins, keratin 5 (KRT5) and 6A (KRT6A) in the keratin gene cluster on chromosome 12 (figure 4A), together with keratin 19 (KRT19), in the equivalent cluster on chromosome 17 are among the most highly expressed genes in the tracheal epithelial cells and all exhibit HTE-selective COREs overlapping their promoters. Moreover, though both KRT5 and KRT6A are expressed in epidermal keratinocytes (NHEKs)7 as well as tracheal epithelial cells, several DHS spanning the KRT5 gene are unique to HTE cells (pink circle in figure 4A). In contrast, the KRT6A promoter exhibits a common DHS in HTE and NHEK cells which is thus lost when the NHEK sites are subtracted (teal circle in figure 4A). One of the critical functions of tracheal epithelial cells is to control the airway luminal environment, so it was probable that we would find some regulatory elements for ion channels. Chloride channel accessory 2 protein (CLCA2), which is known to show some tracheal specificity8 9 and is highly expressed in the cultured HTE cells, is associated with a HTE-selective CORE at its gene promoter, in contrast to the neighbouring CLCA1 gene on chromosome 1, which is expressed in intestinal epithelial cells and lacks an HTE-selective CORE (figure 4B). HTE-selective COREs are also associated with the promoter regions of multiple epithelial transcriptional regulator genes, including ELF3 (epithelial-specific, Ets domain transcription factor, E74-like factor 3),10 Kruppel-like factor 5 (KLF5)11 (see online supplementary figure 2) and GATA-binding protein 6 (GATA6),12 all of which are expressed in these cells and bronchial epithelial cells and/or lung. Several proteins implicated in airway disease also have HTE-selective COREs coincident with their promoters or within the gene itself, including S100 calcium-binding protein A9 (S100A9) which was linked with cystic fibrosis lung disease13–16 and IL2RB which was identified as an asthma susceptibility locus.17 The fact that this gene shows very low expression levels in the HTE cells suggests some of the cis-regulatory elements in the CORE may have an inhibitory effect on gene expression. When we extended this analysis to the list of 974 NCBI reference sequence genes with a greater representation of HTE-selective DHS within their promoters and coding regions than the five background cell types combined (see online supplementary table 1), many of the same loci were identified, including KRT5, KRT6A, KLF5 and S100A9.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
COREs of open chromatin are associated with critical genes in human tracheal epithelial cell function. Each panel (A–C) shows a region of the human genome on the University of California Santa Cruz (UCSC) genome browser (http://genome.ucsc.edu) with combined DNase Solexa (Illumina) sequence data and peak calls for two biological replicates of human tracheal epithelial (HTE) cells and skin fibroblasts (Fibroblasts_park). UCSC genes within the region are also shown. Above these tracks are HTE-selective COREs (horizontal purple bars), HTE-selective DNase I hypersensitive sites (DHS) (vertical purple bars) and (in A, C) HTE-selective sites that do not overlap with sites in normal human epidermal keratinocytes (NHEK) (vertical grey bars, HTE-selective_sites_no_NHEK). Pink and teal circles show HTE-selective COREs or DHS of particular interest (see text). (A) Part of the type II cytokeratin cluster on chromosome 12q12-q13 showing the regions of open chromatin around the keratin 5 (KRT5) and 6A (KRT6A) genes, which are both highly expressed in HTE cells. (B) The calcium-sensitive chloride channel accessory protein (CLCA) cluster on chromosome 1p22. CLCA2 is highly expressed in HTE cells, CLCA1 is not. (C) The EHF–APIP (Ets homologous factor, epithelial specific–APAF1-interacting protein, anti-apoptotic) interval which shows strong association with lung disease severity in a cystic fibrosis modifier genome-wide association study. The pink arrow shows the location of the single nucleotide polymorphism with the highest association. PDHX, pyruvate dehydrogenase complex, component X.
HTE-selective DHS are enriched for binding sites for epithelial transcription factors
The HTE-selective DHS within promoter and intergenic regions were analysed with the use of Clover18 to search for over-represented sequence motifs that could identify transcription factor binding sites used in HTE cells. These sequence regions were each analysed in three different groups: all DHS, HTE-selective DHS and ubiquitous DHS. The results of the promoter analyses are given in online supplementary table 4, and the intergenic analyses are in online supplementary table 5. Of particular interest are the comparisons between the intergenic sites for which the representation of motifs is markedly different in the HTE-selective and ubiquitous sites. As expected, the ubiquitous sites contain a high frequency of CTCF-binding motifs, which are over-represented on 23/23 chromosomes but not in HTE-selective sites. CTCF (CCCTC binding factor) sites are often associated with enhancer blocking insulator function19 20 and play a critical role in maintaining higher order chromatin structure.21–23 In contrast, among HTE-selective sites, binding sites for the epithelial-specific Ets transcription factor ELF5 are over-represented on all chromosomes. ELF5 is known to regulate a number of epithelial-specific genes in tissues containing glandular epithelium.24 Also over-represented in the HTE-selective intergenic sites on multiple chromosomes are binding sites for the Forkhead transcription factors FOXA1 (forkhead box A1, hepatocyte nuclear factor 3α, HNF3α, on 19/23 chromosomes) and FOXA2 (forkhead box A2, HNF3β, on 15/23 chromosomes). These factors are thought to be ‘pioneer’ factors that establish the nucleus of a regulatory complex by opening the chromatin to provide access to other proteins.25 FOXA1/A2 are involved in the development of multiple endoderm-derived organ systems such as lung, pancreas and prostate (reviewed in Kaestner26). Motifs for Kruppel-like factor 4 (KLF4) binding are also over-represented in HTE-selective intergenic DHS on 19/23 chromosomes. KLF4 is known to play a role in epithelial differentiation and to function as both an activator and a repressor.
HTE-selective sites identify enhancer elements in multiple genes
To further validate our genome-wide data in HTE cells we looked for peaks of open chromatin that coincided with previously characterised regulatory elements. Multiple examples were found, including enhancers in the first introns of peptidylarginine deiminase type I27 and aquaporin 528 and 5′ to the keratin 5 gene.29 The location of peaks of open chromatin in HTE cells coinciding with these mapped enhancers are shown in online supplementary figure 3.
HTE-selective sites identify candidate regulatory regions in GWAS
Multiple GWAS have been undertaken to identify novel regions of the genome that contribute to the aetiology of complex lung diseases, such as COPD,30 asthma17 31–33 and sarcoidosis.34 Some of these studies found the strongest associations with single nucleotide polymorphisms (SNPs) located in introns of genes or in intergenic regions, implicating regulatory elements in the mechanism underlying the association. Similarly, a recent GWAS for modifier loci influencing lung disease severity in cystic fibrosis identified several peaks of association with SNPs in non-coding regions.35 Figure 4C illustrates one of these regions of significant association with CF lung severity on chromosome 11p13, between EHF (Ets homologous factor, epithelial specific) and APIP (APAF1-interacting protein, anti-apoptotic) and the location of the SNP, which shows the highest significance (pink arrow). In HTE cells, multiple regions of open chromatin are evident in this genomic region, some of which are HTE specific and others are ubiquitous. Further functional analysis is underway to determine whether these DHS contain regulatory elements such as enhancers, and moreover, which genes are associated with sites that are relevant to cystic fibrosis lung disease severity.
Discussion
A greater understanding of the transcriptional networks that distinguish one differentiated cell type from another will be generated by detailed analysis of regulatory elements genome wide in primary cells. Here we present genome-wide open chromatin data on primary HTE cells generated by DNase-seq. These cells represent the most proximal part of a continuous epithelial sheet that lines the respiratory system from the trachea, through bronchi and bronchioles to the gas exchange surface in alveoli. Some differentiated functions are maintained throughout the epithelium, while others are spatially restricted to different parts of the airway. The generation of additional maps of open chromatin from other primary airway epithelial cell types, such as bronchial cells, will enable bioinformatic comparisons to reveal differences in transcriptional networks and cell-specific regulatory elements in these cell types. This may have significant utility in the clinical management of complex lung disease.
In addition to addressing these more global questions of transcriptional regulation our data provide a valuable resource to search for novel regulatory elements for coordinately regulated gene families and individual genes. One such locus that encodes part of a multi-subunit protein complex and has novel HTE-selective DHS flanking the gene is SCNN1B, which encodes the β subunit of the non-voltage-gated, amiloride-sensitive, epithelial sodium channel. This protein, together with the α and γ subunits encoded by SCNN1A and SCNN1G respectively, is critical to normal fluid transport in the airway epithelium.36–38
Finally, our genome-wide open chromatin data on HTE cells may assist in advancing GWAS for lung diseases, from SNPs associated with a phenotype, to functional elements for mechanistic study. This will be particularly relevant in diseases associated with airway epithelial dysfunction in which critical SNPs are located within non-coding regions of the genome. However, due to the functional complexity of the genome, analysis of these regions should not be restricted to the peaks of open chromatin that coincide with SNPs.
Methods
HTE cells
Human trachea were collected post mortem from healthy donors. HTE cells were isolated from these trachea and grown as described previously.39
DNase-seq
Two technical replicas of DNase-seq were carried out as described previously2 on two independent cultures of primary HTE cells from different donors. Sequencing by Illumina GAIIx produced 39 642 133 and 40 694 564 reads respectively for HTE samples 1 and 2. To check for reproducibility between the samples the percentage of the top 50 000 peaks from sample 1 that overlapped with the top 100 000 peaks from sample 2 were compared, and vice versa. This standard has been accepted by the ENCODE consortium. The overlap was 80% in one direction and 85% in the other direction, which passes the threshold of acceptable reproducibility. The DNase-seq data on five ENCODE cell types (FibroP, GM12878, HeLa-S3, HepG2 and HUVEC) were generated by the ENCODE consortium.
DNase I hypersensitive sites
Full details of the bioinformatic analysis are provided in the online supplement.
Acknowledgments
We are grateful to Dr M Knowles and members of the CF GWAS consortium for sharing data prior to publication; the ENCODE consortium; and Dr K Gaulton for helpful discussions.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
Funding This work was supported by the National Institutes of Health, R01HL094585 (PI:AH), NHGRI U54 HG004363 (PI: GEC) and R01HD068901 (PI:AH).
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.