Article Text

Abstract
Asthma is characterised by airway hyper-responsiveness and remodelling, and there is mounting evidence that alterations in the phenotype of airway smooth muscle (ASM) play a central role in these processes. Although the concept that dysregulation of ASM Ca2+ homeostasis may underlie at least part of these alterations has been around for many years, it is only relatively recently that the availability of ASM biopsies from subjects with mild and moderate asthma has allowed it to be properly investigated. In this article, critical components of the pathobiology of asthmatic ASM, including contractile function, proliferation, cell migration and secretion of proinflammatory cytokines and chemokines, are reviewed and related to associated changes in ASM Ca2+ homeostasis. Based on this evidence, it is proposed that a unifying mechanism for the abnormal asthmatic phenotype is dysregulation of Ca2+ homeostasis caused at least in part by a downregulation in expression and function of sarcoendoplasmic Ca2+ ATPases (SERCAs).
- Airway remodelling
- airway smooth muscle
- asthma
- asthma mechanisms
- calcium
- hyper-responsiveness
- SERCA
Statistics from Altmetric.com
Asthma is a chronic inflammatory disease which is characterised by widespread structural changes in the airways (airway remodelling), encompassing epithelial dysfunction, mucous gland hypertrophy, deposition of extracellular matrix (ECM) and changes in airway smooth muscle (ASM) phenotype.1 ASM from patients with asthma exhibits enhanced contractile activity,2–4 as well as enhanced proliferation, migration and secretion of proinflammatory cytokines.5–8 This article reviews critical components of asthmatic ASM pathobiology and suggests that a unifying mechanism for the abnormal asthmatic phenotype is dysregulation of Ca2+ homeostasis caused at least in part by a downregulation in expression of sarcoendoplasmic Ca2+ ATPases (SERCAs).
Contractility of ASM in asthma
It is widely agreed that alterations in the contractility and dynamics of ASM contribute to airway narrowing and hyper-responsiveness in asthma. The contractile apparatus of ASM is composed of myosin and actin filaments (F-actin), and regulatory protein kinases and phosphatases that control their interaction. Myosin heavy chain (MHC) has ATPase activity, and binds to two light chains including the 20 kDa regulatory light chain (MLC20). F-actin associates with regulatory proteins including tropomyosin, caldesmon and calponin, and promotes myosin assembly and ATPase activity. Contraction is normally initiated by elevation of intracellular [Ca2+] ([Ca2+]i), Ca2+-calmodulin-dependent activation of myosin light chain kinase (MLCK), and consequently phosphorylation of MLC20 and myosin–actin cross-bridge cycling. MLC20 is dephosphorylated by myosin light chain phosphatase (MLCP), which is regulated by Rho kinase, protein kinase C and telokin, so the net phosphorylation state of MLC20 and therefore force production is primarily dependent on the relative activities of MLCK and MLCP9 (figure 1). However, actin-based mechanisms also play an important role through modulation of myosin ATPase activity and cytoskeletal dynamics.10 Pathways that initiate or modulate ASM contraction therefore include those that regulate [Ca2+]i, Ca2+ sensitivity (via MLCP) and the actin cytoskeleton. Alterations to several such pathways have been implicated in asthma.

Basic molecular mechanisms underlying force development in airway smooth muscle (ASM), and their regulation. Force is generated by ATP-dependent interaction between myosin and F-actin, regulated by phosphorylation of myosin 20 kDa regulatory light chain (MLC20) by myosin light chain kinase (MLCK). A rise in [Ca2+]i activates MLCK via Ca2+–calmodulin (CaM). MLC20 is dephosphorylated by myosin light chain phosphatase (MLCP), under the regulation of kinases including Rho kinase and protein kinase C (PKC). Highlighted boxes mark changes reported in asthma or following stimulation with interleukin 13 (IL-13). See text for details.
Increased expression of both a fast MHC isoform and MLCK has been reported in ASM from patients with asthma, which could increase the velocity of ASM shortening.2 4 11 Interestingly, Fairbank et al12 have shown that repeated increases in ASM tone lead to increased MLCK expression, MLC20 phosphorylation and changes in cytoskeletal organisation, suggesting that in asthma these could be secondary phenomena. Rho kinase has also been implicated in the enhanced response of ASM from subjects with asthma,3 though it is unclear whether its expression is altered. Notably, interleukin 13 (IL-13) is reported to increase expression of RhoA, a key activator of Rho kinase, in murine ASM.13
Cytosolic [Ca2+] is dependent on Ca2+ entry and removal across the cell membrane, and sequestration into and release from intracellular stores including the sarcoplasmic reticulum (SR) and mitochondria.14 The highly non-linear relationship between [Ca2+]i and force in smooth muscle (due to 4:1 stoichiometry of Ca2+–calmodulin interaction) implies that even a small subthreshold increase in [Ca2+]i could greatly increase subsequent responses to agonists. It was therefore suggested decades ago that the exaggerated contractile response of ASM typical of asthma could result from abnormal Ca2+ handling.15 This also has implications for mechanisms other than contraction.
Proliferation and migration of ASM in asthma
ASM hyperplasia and proliferation may account for the increased ASM mass characteristic of asthma,5 and have been associated with increased ASM cell division, an altered ECM and reduced apoptosis.16 Cytokines, growth factors and ECM proteins induce ASM hyperplasia, and platelet-derived growth factor (PDGF), epidermal growth factor (EGF), T helper 2 (Th2) cytokines (IL-13 and IL-4), leukotriene D4 (LTD4) and transforming growth factor β (TGFβ) all increase proliferation of ASM in vitro.16 All these mitogens appear to activate the GTPase p21Ras, which in turn activates phosphatidylinositol 3-kinase (PI3 kinase) or extracellular signal-regulated kinase (ERK); both of these regulate ASM proliferation through the cyclin D1 promoter.17 18 Glucocorticoids, a mainstay of asthma treatment, suppress proliferation in part via decreasing cyclin D1 expression,19 mediated by a complex of the glucocorticoid receptor and CCAAT/enhancer-binding protein α (C/EBPα). However, ASM from subjects with asthma exhibits a tissue-specific lack of C/EBPα, and this has been postulated to confer an ASM phenotype that is more susceptible to mitogenic and contractile stimuli.20
Migration of proliferating ASM and precursor cells may contribute to increased ASM mass in asthma, and involves reorganisation of the actin cytoskeleton, assembly of focal contacts and myosin–actin-mediated force generation. Many factors integrate to promote actin disassembly, polymerisation and attachment to focal adhesions, and regulation of myosin activity; several are also involved in ASM growth (eg, PI3 kinase and ERK) and contraction (eg, MLCK, MLCP, RhoA and Rho kinase) (see review by Gerthoffer21). While the role of cell migration in airway remodelling remains unclear, ASM cells derived from subjects with asthma exhibit enhanced migration.6 8 Migration is strongly Ca2+ dependent, as myosin activity is regulated by Ca2+–calmodulin-dependent activation of MLCK, and reorganisation of the actin cytoskeleton involves several Ca2+-dependent mechanisms, notably the actin-severing protein gelsolin.21
Secretion of cytokines and chemokines in asthma
ASM is a rich source of chemokines and cytokines, including eotaxin, RANTES (regulated upon activation, normal T cell expressed and secreted), TARC (thymus and activation-regulated chemokine), SCF (stem cell factor) and GM-CSF (granulocyte–macrophage colony-stimulating factor).22 For example, eotaxin release following stimulation of ASM with IL-13 or IL-4 mediates eosinophil recruitment,23 and ASM-derived RANTES induces chemotaxis of inflammatory cells, thereby contributing to the inflammatory process.24 Ca2+ signalling has an important role in release of cytokines and chemokines (eg, Denecker et al25 26), and the finding that IL-13-induced eotaxin-1 secretion from healthy ASM is enhanced by knockdown of SERCA2 also implicates a key role for [Ca2+]i.8
Ca2+ homeostasis in ASM
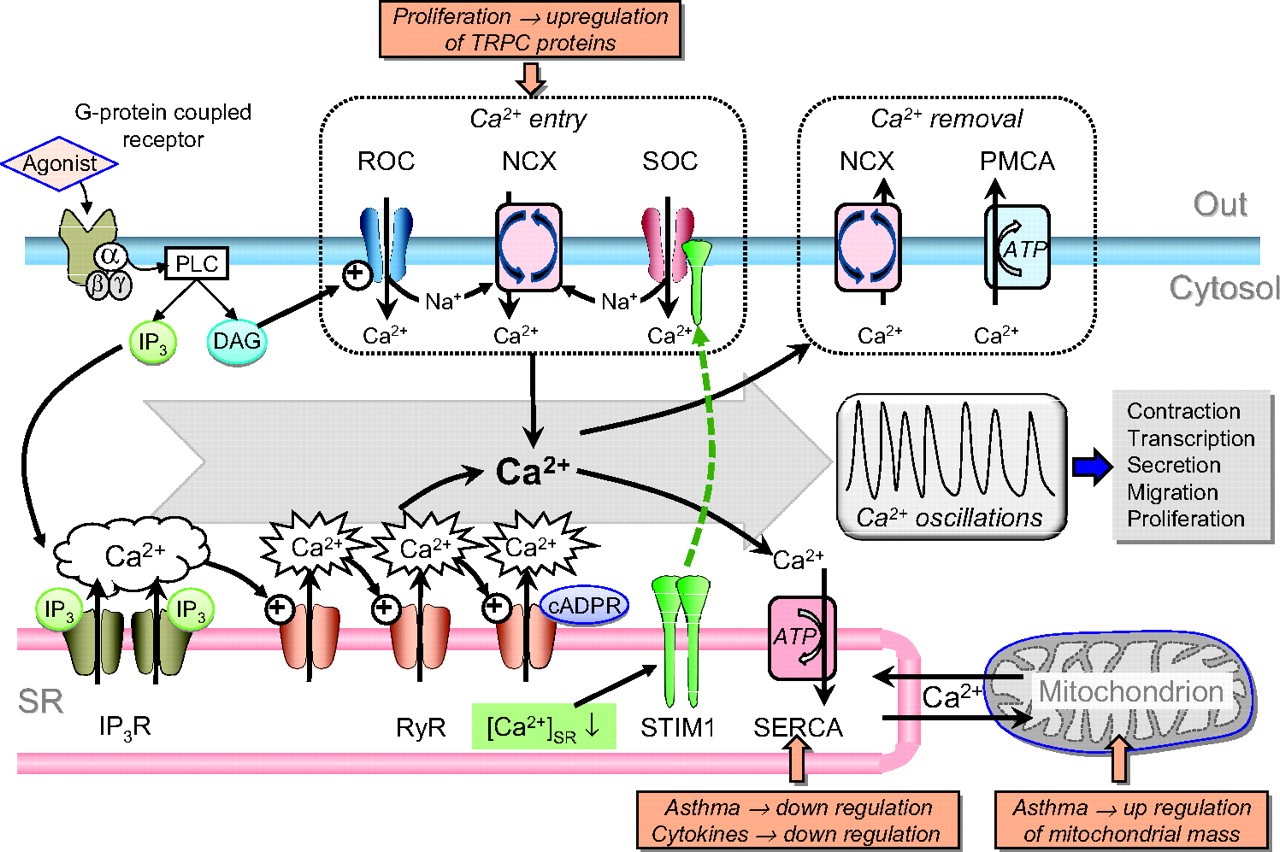
Intracellular Ca2+ has a central role in transduction of signals leading to diverse cellular responses, including contraction, metabolism, cell motility and protein secretion, as well as cell differentiation and proliferation.14 21 27 As such, it has long been suggested that alterations in ASM Ca2+ homeostasis may be central to the development of the asthmatic ASM phenotype and hyper-responsiveness.15 28 29 Elevation of ASM [Ca2+]i can be elicited by activation of Ca2+ influx pathways and/or release from the SR. Ca2+ then binds to and stimulates high affinity binding proteins such as calmodulin, which activates MLCK,14 and the phosphatase calcineurin, which activates the transcription factor NFAT (nuclear factor of activated T cells) and indirectly NF-κB (nuclear factor-κB), both of which are implicated in inflammation and airway remodelling in asthma.30 31 [Ca2+]i is pumped back into the SR or out of the cell in order to terminate its effects, the former mediated by the SERCA, and the latter by the plasma membrane Ca2+ ATPase (PMCA) and Na+/Ca2+ exchanger (NCX)14 (figure 2).

Ca2+ mobilisation pathways in airway smooth muscle (ASM). Binding of an agonist to a G-protein-coupled receptor activates phospholipase C (PLC) to generate diacylgycerol (DAG) and inositol trisphosphate (IP3). DAG activates receptor-operated channels (ROCs), eliciting Na+ and Ca2+ entry, while IP3 activates clusters of IP3 receptors (IP3Rs) on the sarcoplasmic reticulum (SR), causing Ca2+ release which may activate adjacent ryanodine receptors (RyRs) to increase the frequency of sparks. RyRs may also be activated or potentiated by cADP ribose (cADPR). Store depletion is sensed by stromal-interacting molecule 1 (STIM1) within the SR, which translocates to and activates store-operated channels (SOCs). The elevation in subplasmalemmal [Na+] resulting from activation of non-selective ROCs or SOCs may be sufficient to drive reverse-mode operation of Na+/Ca2+ exchanger (NCX), leading to Ca2+ entry. Much of the Ca2+ entering the cell and released from stores may be sequestered by the superficial SR through sarcoendoplasmic Ca2+ ATPases (SERCAs) and mitochondria. Cycling between SR Ca2+ uptake and release mechanisms leads to Ca2+ oscillations. Ca2+ is removed from the cell by the plasma membrane Ca2+ ATPase (PMCA) and normal operation of the NCX. TRPC, transient receptor potential channel.
Agonists binding to Gq/G11 protein- or tyrosine kinase-coupled receptors can initiate elevation of [Ca2+]i via phospholipase C (PLC)-mediated generation of diacylglycerol (DAG) and inositol trisphosphate (IP3). DAG activates non-selective cation channels (permeable to both Na+ and Ca2+), often referred to as receptor-operated channels (ROCs)29 32; these may be formed of TRPC6 (transient receptor potential channel 6) proteins which are expressed in human ASM.33 IP3 binds to clusters of receptors (IP3Rs) in the SR to initiate Ca2+ release and highly localised elevations in [Ca2+] (‘puffs’), which consequently activate ryanodine receptors (RyRs) in the SR (Ca2+-induced Ca2+ release, CICR), thereby amplifying the elevation in cytosolic [Ca2+] and leading to Ca2+ ‘sparks’, and potentially regenerative Ca2+ waves and oscillations.14 RyRs are also activated by cADP ribose (cADPR), which is synthesised by the multifunctional enzyme CD38 (an ADP-ribosyl cyclase) and may contribute to generation of Ca2+ sparks and waves; it has been proposed that this pathway plays an important role in ASM.34 It has been reported that some agonists such as LTD4 do not elicit IP3-mediated Ca2+ release in human bronchioles, but do activate Ca2+ entry via ROCs.32
Depletion of SR Ca2+ content by the above processes activates store-operated Ca2+ entry (SOCE) via the SR Ca2+-sensing protein stromal-interacting molecule 1 (STIM1), which associates with the plasmalemmal protein Orai causing it to form channels which mediate SOCE.35 36 In smooth muscle many studies suggest that the channels underlying SOCE (store-operated channels; SOCs) are non-selective (ie, Na+ and Ca2+ permeable), unlike classical STIM1–Orai which is Ca2+ selective. Possibly explaining this, there is evidence that the STIM1–Orai complex also incorporates TRPC proteins in smooth muscle, although this remains controversial.37 As the degree of SR Ca2+ loading regulates SOCE, SERCA activity directly influences Ca2+ influx.
Removal of Ca2+ from the smooth muscle cell is handled by PMCA and NCX. PMCA is an ATP-dependent transporter activated by protein kinase G and A,14 providing one mechanism by which agents such as β2-adrenoceptor agonists cause ASM relaxation. NCX uses the Na+ electrochemical gradient to pump out 1 Ca2+ in exchange for 3 Na+, and as such is dependent on membrane potential and cytosolic [Na+]. Elevation of subsarcolemmal [Na+] can therefore cause reverse-mode operation of NCX and Ca2+ entry, and there is evidence that in ASM agonist-induced Na+ entry through non-selective cation channels (see above) does just this, with NCX then contributing to the elevation of [Ca2+]i and store refilling.38 39
The intracellular structure of ASM is highly organised, with peripheral SR, mitochondria and plasmalemma closely approximated in localised signalling microdomains. This provides a ‘superficial buffer barrier’, which buffers Ca2+ entry and limits its pervasion into the deeper cytosol, and through its highly regulated Ca2+ release and uptake mechanisms provides a level of precise control over the spatial and temporal characteristics of changes in [Ca2+]i.28 40 As such, it facilitates a high level of cross-talk between these organelles and consequently the mechanisms discussed above, and thus the ability to engender regenerating Ca2+ waves and oscillations. In ASM, agonists have indeed been shown to evoke Ca2+ waves or sustained oscillations of [Ca2+]i, caused by sequential Ca2+ release from and re-uptake into the SR, involving IP3Rs, RyRs and SERCAs29 41 42 (figure 2). These waves seem to be responsible for acetylcholine-mediated contraction in human ASM, since their suppression by blockade of SERCAs or RyRs causes relaxation.41 Ca2+ entry is nevertheless important, as wave activity is terminated and contraction strongly suppressed by blockade of non-selective cation channels (ie, SOCs and ROCs) and reverse-mode NCX, although inhibition of L-type Ca2+ channels has only a minor effect.29 41 This implies that Ca2+ entry is primarily voltage independent and funnelled into the SR rather than serving to elevate [Ca2+]i directly. Importantly, Ca2+ waves and oscillations also activate other Ca2+-dependent processes, including gene transcription.29 43
SERCAs clearly play a critical role in the above processes. There are three tissue-specific members of the mammalian SERCA family, SERCA1, SERCA2 and SERCA3, of which SERCA2 is expressed in muscle cells. Alternative splicing of exon 20 of the ATP2A2 gene gives rise to SERCA2A, SERCA2B and SERCA2C mRNA transcripts, which differ in the final exon usage.44 We have recently demonstrated that the predominant isoform in ASM is SERCA2B, with the other splice variants expressed at very low levels.8 SERCAs are regulated by phospholamban, which under basal conditions acts to inhibit itheir activity. When phospholamban is phosphorylated, for example by protein kinase A, this inhibition is relieved and SR Ca2+ uptake dramatically increased.45 This provides another important mechanism by which cAMP-raising agents induce bronchial relaxation.
Altered ASM Ca2+ homeostasis in asthma: role of SERCAs
We have recently reported that SERCA2 protein expression in both native and cultured ASM from endobronchial biopsies of patients with mild and moderate/severe asthma is diminished compared with that of healthy subjects; the extent of this effect correlated with disease severity.8 Agonist-induced Ca2+ release from the SR was similarly depressed, consistent with a reduced store content, and the rate at which bradykinin-induced elevation of [Ca2+]i recovered to baseline levels was significantly slowed (figure 3). In addition, ASM cells from subjects with asthma were significantly more active in a Ca2+-dependent spreading assay that reflects migration. Importantly, suppression of SERCA2 by small interfering RNA (siRNA) in ASM cells derived from donors without asthma recapitulated the asthmatic phenotype, with similarly slowed recovery of [Ca2+]i to baseline and increased rates of proliferation and cell spreading. Moreover, IL-13-induced eotaxin release, which is greatly enhanced in cultured ASM cells from subjects with moderate/severe asthma, was also increased by siRNA-induced SERCA2 knockdown.8

{kind=link}
{kind=link}
{kind=link}
Recovery of Ca2+ transient is delayed in airway smooth muscle (ASM) cells from subjects with asthma. (A) An example trace of a Ca2+ transient elicited by 1 μM bradykinin in ASM cells cultured from a healthy subject, measured using Fura2. The time taken for the increase in Ca2+ to return to baseline is indicated. (B) Mean data obtained from ASM cells derived from healthy subjects and those with mild and moderate asthma (data from Mahn et al8). *p<0.05.
The above strongly suggests that downregulation of SERCA2 plays an important, if not critical role in the dysregulation of ASM Ca2+ homeostasis in asthma and the consequent alterations in phenotype. Importantly, Sathish et al46 have now reported that treatment of healthy ASM with the proinflammatory cytokines IL-13 and tumour necrosis factor α (TNFα) causes suppression of SERCA2 expression and function, thereby providing a putative link between airway inflammation and reduced ASM SERCA2 expression in asthma. Interestingly, IL-13- and TNFα-induced hyper-responsiveness has also been associated with altered SR Ca2+ handling via the CD38/cADPR pathway.47 48
It is noteworthy that a fall in expression of SERCA2A/2B occurs in Darier disease, an autosomal dominant skin condition in which only one copy of the SERCA2 gene (ATP2A2) is functional. This disease is characterised by lesions demonstrating abnormal keratinisation and loss of epidermal cellular adhesion,49 though no association with asthma has been reported. Similarly ATP2A2 heterozygous knockout mice eventually develop hyperkeratinised squamous cell tumours.50 These mice also show a deficient cardiac relaxation, and it is known that SERCA2 expression is reduced in human cardiac failure and hypertrophy51; notably, SERCA2 gene transfer into the myocardium is currently undergoing clinical trials for treatment of this condition.
It is not yet known whether the decrease in ASM SERCA2 expression in asthma could have a genetic basis, in which case it might be expected that downregulation of SERCA2 would be observed in other types of cell. Although asthma has not been associated with cardiac dysfunction, which might be predicted if SERCA2 expression is also reduced in cardiac myocytes, patients with Darier disease also do not exhibit any abnormalities of cardiac function.52 Thus even this substantial genetically determined deficit in SERCA2 can cause abnormalities which manifest only in certain types of cell. It may therefore be that mutations in the ATP2A2 gene, or in genes which affect its expression, exist in those with asthma, but for some reason exert restricted effects. This would be analogous to the situation in familial pulmonary arterial hypertension, which is associated with the decreased expression of the BMPR-II receptor caused by autosomal dominant mutations in the BMPR2 gene.53 However, this mutation has a penetrance of only ∼20% and, even though this receptor is functionally important in other organs, causes a lung-specific pathology. This implies that the disease only manifests itself when other factors are present (‘second hit’ hypothesis). It is, for example, feasible that a localised functional suppression of SERCA2 activity may also occur in the powerfully oxidising and cytokine-rich milieu of the inflamed asthmatic lung54 which could constitute a focal ‘second hit’ sufficient to cause a significant effect on cellular Ca2+ handling restricted mainly to these cells. Interestingly, it has recently been reported that SERCA function is inhibited by ORMDL3, which is implicated in the inflammation-promoting unfolded protein response and has been identified as a genetic risk factor for childhood asthma.55 This is of clear relevance to whether the observed reduction in ASM SERCA2 expression is of genetic origin or secondary to an increase in proinflammatory cytokines.46 It is noteworthy that the disease severity-related fall in SERCA2B expression persisted in ASM cells through multiple passages in culture,8 suggesting either a genetic or an epigenetic origin.
Downregulation of SERCA2 function has previously been reported to promote proliferation in a number of tissues including other smooth muscles, possibly mediated via calcineurin.27 It is noteworthy in this regard that ciclosporin, a calcineurin antagonist, has been used as a treatment for steroid-resistant asthma.56 Trian et al57 have proposed that the enhanced proliferation in ASM from subjects with asthma is due to an increase in mitochondrial mass, driven by upregulation of mitochondrial transcription factors as a consequence of altered ASM Ca2+ homeostasis. While it is unclear how increased mitochondria might promote proliferation, this mechanism could be an important downstream effector pathway for SERCA dysfunction, although Trian et al57 advocated an enhanced Ca2+ entry pathway. Notably, a variety of Ca2+ influx pathways are also upregulated when SERCA expression or function is suppressed, for example NCX and TRPC proteins in cardiomyocytes, again associated with an increase in transcript levels of a calcineurin-dependent transcription factor complex and activity of NFAT.58 59 It is therefore interesting that ASM proliferation has been associated with a marked upregulation of TRPCs.60
As SERCA2 plays a key role in the operation of the superficial buffer barrier and generation of Ca2+ oscillations,14 28 40 it can be predicted that reduced expression and/or function of SERCA2 would result in not only increased basal levels of [Ca2+]i, but also potentiation of Ca2+ influx-induced elevations of [Ca2+]i and altered dynamics of Ca2+ oscillations. Consistent with this, an increased resting [Ca2+]i and slowing of Ca2+ oscillations has been observed in ASM under conditions where SERCA activity is reduced.46 This could underpin, at least in part, both ASM hyper-responsiveness to agonists and, through alterations to the frequency of Ca2+ oscillations, differential activation of transcription factors such as NFAT and NF-κB.43
Conclusion
It is clear that Ca2+ signalling plays a central and critical role in most if not all of the cellular processes that are known to be altered in ASM from subjects with asthma, including contractility, responsiveness to agonists, proliferation, migration and secretion of inflammatory mediators. We therefore propose that a unifying mechanism for the abnormal asthmatic phenotype is dysregulation of Ca2+ homeostasis, mediated at least in part by downregulation of SERCA2 expression and function driven by proinflammatory mediators. Many questions remain unanswered, including whether the reduction in SERCA2 expression is induced or of genetic origin, and whether observed changes in other Ca2+ homeostatic mechanisms occur independently or are secondary to reduced SERCA2 function. However, the fact that experimental suppression of SERCA2 expression in healthy ASM appears to recapitulate key aspects of the asthmatic phenotype8 strongly suggests that SERCA2 downregulation plays a central role in the altered function of asthmatic ASM.
Acknowledgments
Work in the Authors' laboratories was supported by Asthma UK, MRC, Wellcome Trust and the NIHR Guy's and St Thomas' Foundation Trust/KCL Biomedical Research Centre. None of the authors have competing interests.
References
Footnotes
Funding Asthma UK, MRC, NIHR Guy's and St Thomas' Foundation Trust/KCL Biomedical Research Centre. Other Funders: Wellcome Trust.
Competing interests None.
Ethics approval This study was conducted with the approval of the Guy's and St Thomas' Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.