Article Text

Abstract
Background: Activated protein C (APC) significantly decreases mortality in severe sepsis, but its role in acute lung injury from non-infectious aetiologies is unclear. The role of APC in hyperoxic acute lung injury was tested by studying the physiology of lung injury development, measurement of key coagulation proteins and treatment with murine APC (mAPC).
Methods: Mice were continuously exposed to >95% oxygen and lung injury was assessed by extravascular lung water, lung vascular protein permeability and alveolar fluid clearance. Coagulation proteins were measured in bronchoalveolar lavage (BAL) fluid and plasma. Recombinant mAPC was administered in preventive and treatment strategies.
Results: Hyperoxia produced dramatic increases in lung vascular permeability and extravascular lung water between 72 and 96 h. Lung fluid balance was also adversely affected by progressive decreases in basal and cAMP-stimulated alveolar fluid clearance. Plasma levels of APC decreased at 72 h and were 90% depleted at 96 h. There were significant increases in BAL fluid levels of thrombomodulin, thrombin-antithrombin complexes and plasminogen activator inhibitor-1 at later time points of hyperoxia. Lung thrombomodulin expression was severely decreased during late hyperoxia and plasma levels of APC were not restored by excess thrombin administration. Administration of recombinant mAPC failed to improve indices of lung injury.
Conclusions: Hyperoxic acute lung injury produces procoagulant changes in the lung with a decrease in plasma levels of APC due to significant endothelial dysfunction. Replacement of mAPC failed to improve lung injury.
Statistics from Altmetric.com
There have been several recent advances in the management of patients with acute lung injury (ALI),1 2 but trials of pharmacotherapies targeting disease pathogenesis have been disappointing.3 The management of sepsis, which is a major cause of ALI and shares many pathophysiological similarities, has been aided by the treatment of patients with activated protein C (APC).4 This pharmacotherapy significantly reduces mortality in selected patients with severe sepsis. The mechanism of this benefit has not been clearly elucidated, especially since APC has many potential beneficial properties including its anticoagulant, anti-inflammatory, anti-apoptotic and endothelial barrier-enhancing properties.5
Sepsis is characterised by a robust inflammatory response and also by activation of the coagulation pathways. The pathogenesis of ALI is similar to sepsis, with exuberant inflammation and a procoagulant antifibrinolytic environment in the alveolus.6 7 The normal uninjured alveolar epithelium potentially favours anticoagulation, and it has recently been reported that it contains thrombomodulin (TM) and endothelial protein C receptor (EPCR) and can activate protein C.8 ALI is characterised by low plasma and pulmonary oedema fluid protein C levels9 and high plasma and pulmonary oedema fluid levels of plasminogen activator inhibitor-1 (PAI-1) and TM.9 10 It is therefore reasonable to hypothesise that treatment with APC could be beneficial in patients with ALI.
Because much has already been learned about the coagulation response in infectious models of ALI, we chose to investigate a standard non-infectious model of ALI—namely, severe hyperoxia. The coagulation response in hyperoxia has been incompletely characterised. Gene expression profiling in early hyperoxia has revealed a decrease in TM expression that has been confirmed at the protein level.11 In addition, fibrin deposition in the lungs of mice at later time points of hyperoxia is prominent, potentially indicating a procoagulant microenvironment in the alveolus.12 Also, PAI-1 knockout mice have a significant survival advantage over wild-type controls, suggesting that impaired fibrinolysis is important in the pathogenesis of hyperoxic lung injury.12
There were three main objectives for these studies in hyperoxia. First, in order to place the coagulation response to hyperoxia in the appropriate context, we fully characterised the dynamics of lung injury development secondary to hyperoxia in mice. Second, we measured endogenous plasma levels of murine APC (mAPC) in addition to plasma and bronchoalveolar lavage (BAL) fluid levels of key coagulation and fibrinolysis proteins. We also conducted thrombin challenge experiments to help determine the mechanism of coagulation derangements with hyperoxia. Finally, we tested recombinant mAPC in prevention and treatment strategies in animals exposed to hyperoxia.
METHODS
Hyperoxia model
Male 8–12-week-old C57Bl/6 mice (Jackson Laboratories) were placed in a sealed plexiglass chamber through which 100% oxygen was continuously flowing at 2–3 l/min. An oxygen sensor (Telldyne) was used to maintain oxygen saturations of ⩾95%. Carbon dioxide levels were measured and were <0.2% on repeated measurements. Room air control animals were placed in the plexiglass chamber and exposed to 21% oxygen at 2–3 l/min. Mice had access to standard chow and water at all times. Animal protocols were approved by the University of California, San Francisco Animal Care Committee.
Quantification of lung injury
The gravimetric method was used to measure extravascular lung water and injected 125I-albumin (Iso-Tex Diagnostics) intraperitoneally to measure lung vascular permeability to protein, as described elsewhere.13–15
Alveolar fluid clearance experiments
As previously described,14 15 an in situ model was used to measure net alveolar fluid clearance (AFC) under basal and cAMP-stimulated conditions (isoproterenol 10−4 M).
Blood and BAL fluid collection and measurement of coagulation markers
Mice were euthanised with intraperitoneal pentobarbital and blood was withdrawn by cardiac puncture into syringes containing citrate (0.105 M) and benzamidine (20 mM final). After tracheotomy and insertion of a 20 gauge angiocath, 1 ml of cold phosphate-buffered saline (PBS) containing 10 mM citrate and 20 mM benzamidine (pH 7.4) was lavaged three times into the mouse lungs. BAL fluid total protein was measured spectrophotometrically using Bio-Rad protein reagent. Standard ELISAs were used to measure thrombin-antithrombin (TAT) complexes (Enzygnost, DadeBehring) and PAI-1 (Molecular Innovations). Plasma protein C and soluble EPCR were measured as previously described.16 17 An enzyme capture assay for mAPC was done in the laboratory of Dr Charles T Esmon, as previously described.16 A mouse TM assay was developed using an anti-mouse TM monoclonal antibody (#1699, Esmon laboratory), recombinant mouse TM (Esmon laboratory) and a biotinylated goat anti-mouse TM polyclonal antibody (#409, Esmon laboratory).
APC in vivo studies
Recombinant mAPC was produced in the laboratory of Dr Esmon and APC was assayed as previously described.16 mAPC was administered by the following routes: (1) intraperitoneal, (2) subcutaneous by continuous osmotic pump delivery, (3) intratracheal and (4) intravenous. For the intratracheal route we used a method of direct visualisation and instillation as previously described.18 Human APC (Sigma) was also used in selected experiments.
Intravenous thrombin studies
To determine the effect of excess exogenous thrombin on mAPC generation, murine thrombin (Sigma, 10 units/kg) was administered via the jugular vein to anaesthetised mice. Normal saline was used in the control group. After 3 min the mice were euthanised and blood was withdrawn as described above.
Histology and immunohistochemistry
Mouse lungs were inflated and immersed in 4% paraformaldehyde and embedded in paraffin for routine H&E histological examination. Paraffin-embedded mouse lung tissue (5 μm sections) was also processed for routine immunohistochemistry. After antigen retrieval, a biotinylated goat anti-mouse TM polyclonal antibody (#409, Esmon laboratory) was used for mouse TM immunohistochemistry.
Statistical analysis
Data are presented as mean (SD). The Student t test was used for comparison of two groups or time points and ANOVA with post-hoc Bonferroni correction was used for multiple comparisons (SPSS Version 16.0). For all analyses a p value of <0.05 was considered statistically significant.
RESULTS
Physiological studies of lung fluid balance in hyperoxic lung injury
In order to fully understand the coagulation response to hyperoxia, we first quantified the lung injury response at the major time points of hyperoxic exposure. A clear phenotypic response to hyperoxia occurred after approximately 48 h of exposure with decreased activity and eating as well as huddling and hypothermia of mice. We also observed an LD50 at approximately 96 h of exposure. We therefore chose the following experimental time points to study: 0, 48, 72 and 96 h.
Figure 1A shows that, in parallel to the behavioural changes in the mice, there was a trend for an increase in lung vascular permeability to protein at 48 h of exposure without a significant increase in pulmonary oedema at this time point. At 72 h, both extravascular lung water (fig 1B) and lung vascular protein permeability were significantly increased, with an exponential increase between the 72 and 96 h time points. At 96 h, on post-mortem examination, frank oedema fluid was often apparent in the major airways of the lungs.
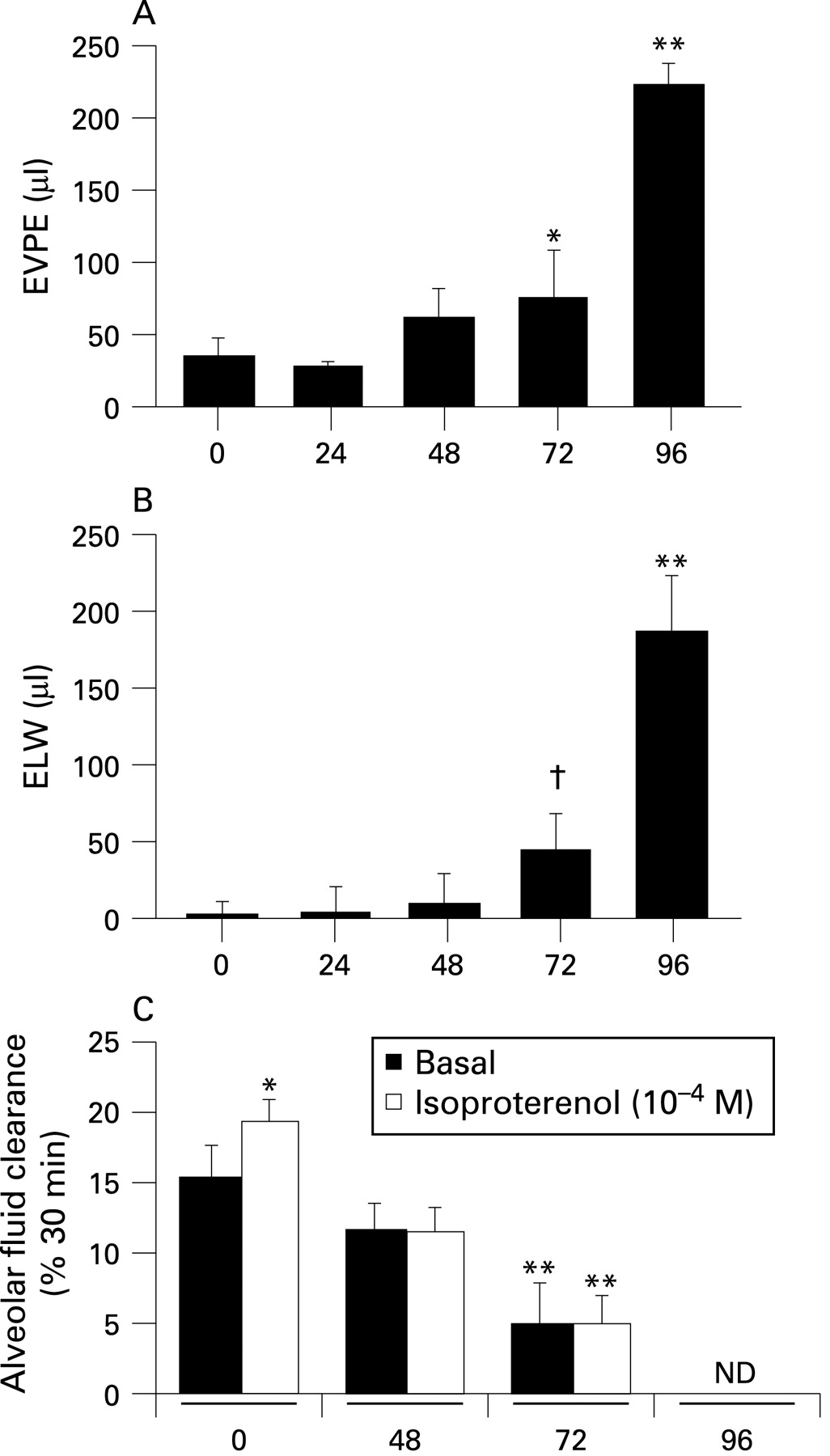
Figure 1C shows the results of both basal and cAMP-stimulated AFC with hyperoxia exposure. Basal AFC first significantly decreased at 72 h and AFC was completely absent at 96 h. Notably, the cAMP augmentation of AFC at baseline was lost at the relatively early 48 h time point. The increase in extravascular lung water (fig 1B) was thus related to increased lung vascular permeability (fig 1A) and a progressive decrease in the clearance capacity of the alveolar epithelium. Figure 1S (in the online supplement) shows the histological correlation of these physiological measurements. At 72 h of hyperoxia there was significant interstitial oedema (Figure 1S:C) that progressed to frank alveolar oedema by 96 h (Figure 1S:D).
Coagulation assessment in hyperoxic lung injury
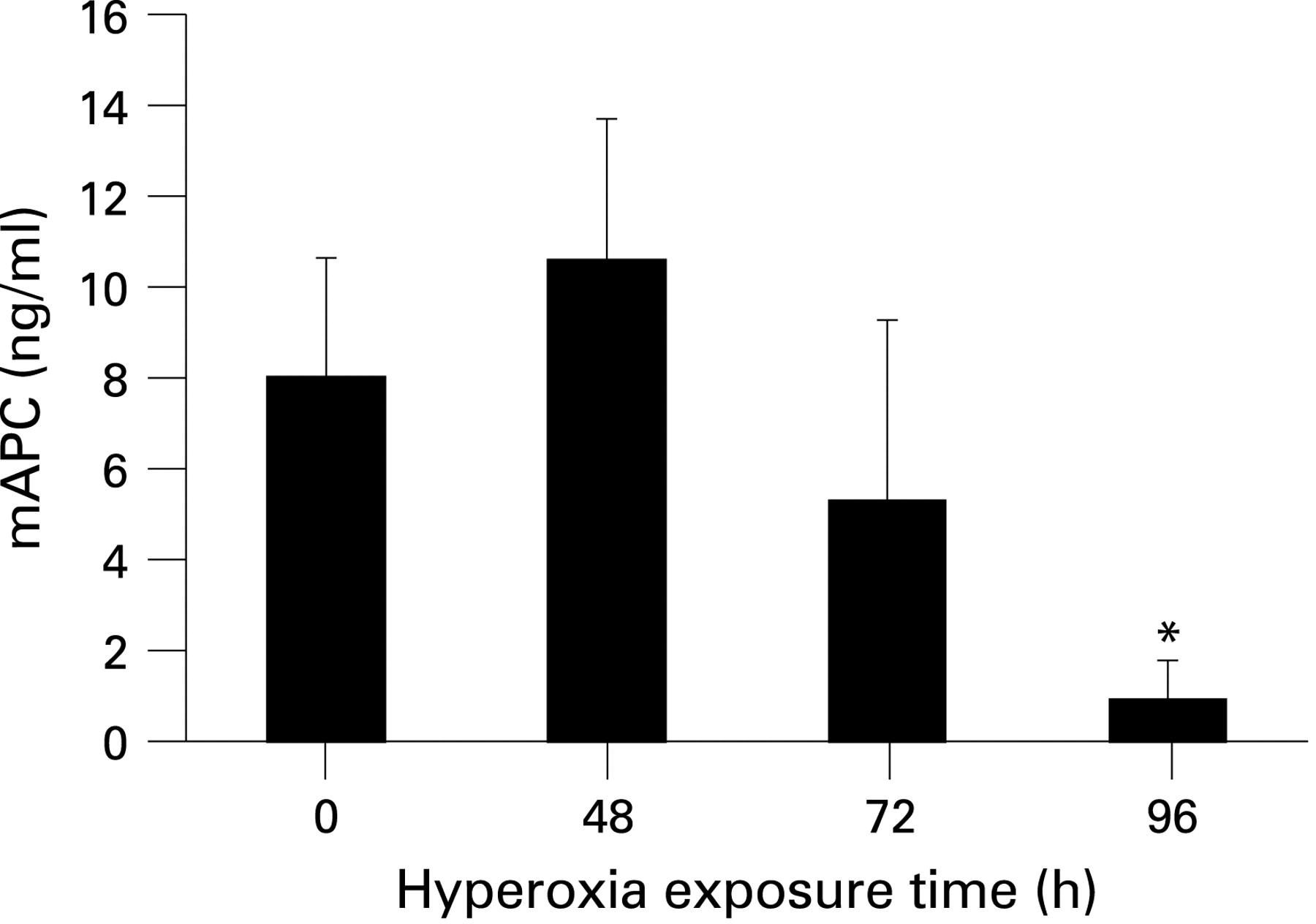
Plasma levels of mAPC were measured by an enzyme capture assay at the previous hyperoxia time points. In mice exposed to room air, mAPC plasma levels were 8–10 ng/ml with a trend for decreased levels at 72 h and significantly decreased plasma levels at 96 h (fig 2). Plasma levels of other coagulation markers were also measured at these same time points. Mouse TM was detectable at baseline, followed by a decrease at 48 and 72 h and then an increase back to baseline plasma levels at 96 h (Figure 2S:A in the online supplement). Plasma levels of TAT, PAI-1, protein C and soluble EPCR were not significantly changed with hyperoxia, except for small increases in soluble EPCR at 48 h and PAI-1 at 72 h, both of which returned to baseline levels at 96 h (Figure 2S:B–E in online supplement). However, there were significant increases in BAL fluid levels of total protein, TM, TAT and PAI-1 at 72 and 84 h of hyperoxia (fig 3A–D). BAL fluid levels of TAT also increased at 48 h of hyperoxia (fig 3C). BAL fluid levels of mAPC were essentially undetectable at all time points (fig 3E).


To determine the mechanism of mAPC depletion with hyperoxic lung injury, excess murine thrombin was injected intravenously to mice at later time points of hyperoxia. Our hypothesis was that the molecular machinery needed to generated mAPC (TM, EPCR) was missing from the severely damaged lung endothelium. If this was correct, then excess thrombin should not be able to rescue mAPC plasma levels. With thrombin challenge (10 units/kg intravenously) there were small non-statistically significant increases in plasma mAPC levels at 72 and 84 h of hyperoxia, with the thrombin-stimulated mAPC levels at 72 h reaching room air (control) concentrations (fig 3S in online supplement). However, at 84 h of hyperoxia, plasma levels of thrombin-stimulated mAPC remained well below control values. Mice were too sick at the 96 h time point for these thrombin challenge experiments.
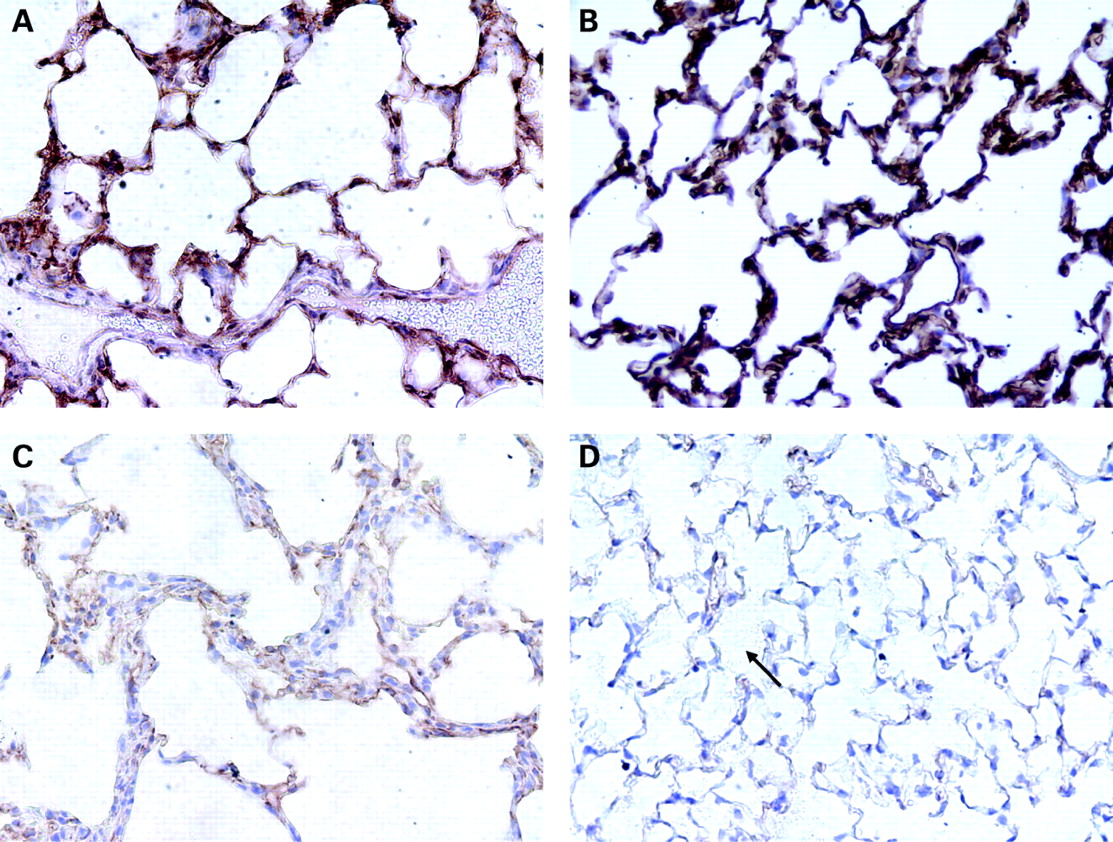
Given the low mAPC plasma levels and the inability of excess thrombin to fully restore mAPC levels, we hypothesised that TM and/or EPCR expression on the lung endothelium were significantly decreased with hyperoxic lung injury. In fact, fig 4 shows that lung TM staining mirrors the time course of mAPC plasma levels in hyperoxia. At 48 h there appears to be an increase in TM staining with a significant decrease in staining intensity at 72 h and a further decrease at 84 h.

Replacement therapy with mAPC
We tested the potential therapeutic significance of the decrease in mAPC plasma levels on the course of hyperoxic lung injury by treating the mice with recombinant mAPC. There was no significant plasma absorption from either the intraperitoneal or subcutaneous (including osmotic pump delivery) routes of delivery. Bolus intravenous delivery provided only transiently increased plasma levels of mAPC due to the plasma half-life in mice of approximately 2 min. Since a previous investigation of human APC in bleomycin lung injury used the intratracheal route of delivery,19 we also tested this route with mAPC . In fig 5, mice were given 1 mg/kg mAPC intratracheally and BAL was performed at 60 min and 6 h. Approximately 680 ng/ml of mAPC was recovered at 60 min, decreasing to 40 ng/ml at 6 h. Plasma levels of mAPC at these corresponding time points were not significantly changed. BAL fluid and plasma levels of mAPC were also measured in mice given mAPC (1 mg/kg intratracheally) at 48 h of hyperoxia and then killed 24 h later. BAL fluid levels of mAPC remained at 40 ng/ml (fig 5A), while plasma levels of mAPC significantly increased to approximately 25 ng/ml (fig 5B), which represents a threefold increase from baseline (room air) concentrations. Given the limitations of continuous intravenous delivery in mice, we chose the intratracheal route for our prevention and treatment experiments.

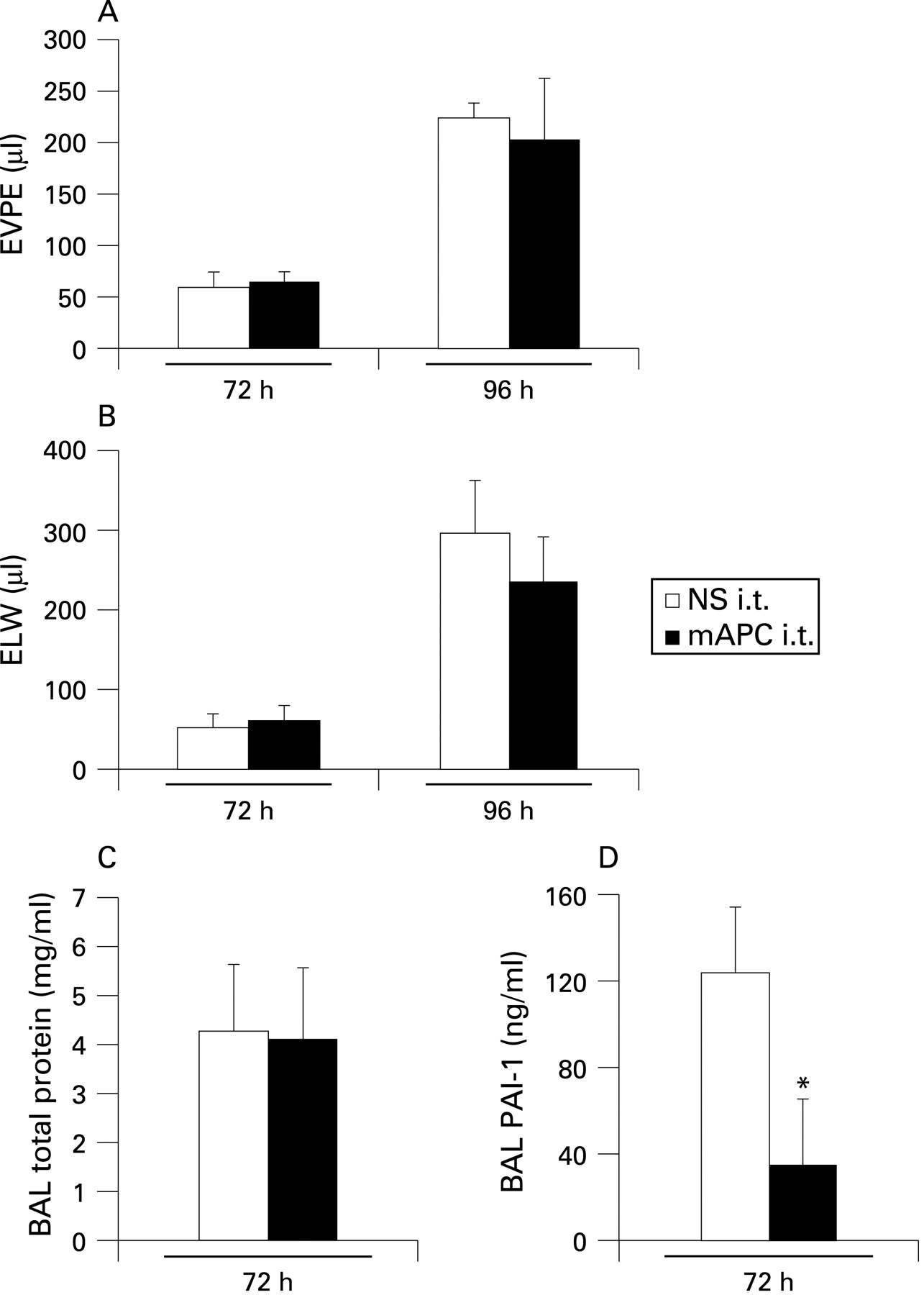
Mice were anaesthetised and given daily intratracheal doses of mAPC (1 mg/kg) in both prevention and treatment strategies. Figure 6A–B shows the results from a prevention strategy at 72 and 96 h of hyperoxia, which showed no significant effect from daily mAPC (intratracheal) compared with control. When mice were given intratracheal mAPC in a treatment strategy starting at 48 h (fig 6C and D) there was no difference in lung injury (BAL total protein, fig 6C) at 72 h of hyperoxia. However, the intratracheal mAPC did have a biological effect by lowering BAL fluid levels of PAI-1 at 72 h (fig 6D). We also tested intratracheal human APC (Sigma, 1 mg/kg) in treatment (n = 5) and prevention (n = 5) strategies without any change in lung injury compared with controls (data not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DISCUSSION
The three major findings of this study are (1) hyperoxia causes time-dependent increases in permeability pulmonary oedema with a concomitant loss of alveolar fluid clearance mechanisms; (2) mAPC plasma levels significantly decrease with moderate to severe hyperoxic lung injury due to severe endothelial dysfunction; and (3) despite procoagulant changes in the lung with hyperoxia, recombinant mAPC replacement via the intrapulmonary route does not ameliorate hyperoxic lung injury.
In order to accurately characterise the coagulation response to hyperoxia, we first fully assessed the time course of lung injury development, an issue that has not previously been reported in a single investigation. The results showed that both the alveolar epithelium and the lung vascular endothelium are dysfunctional as early as 48 h of hyperoxia. At 72 h of hyperoxia, permeability pulmonary oedema is evident and is accompanied by a significant impairment in alveolar fluid clearance. By the 96 h time point, alveolar epithelial fluid transport has been completely lost which, in combination with a progressive increase in vascular permeability, results in death.
We also determined the coagulation response to hyperoxia locally in the lung and in the plasma compartment. At later time points of hyperoxia there were robust increases in TAT, TM and PAI-1 in the lung, implying a significant procoagulant environment. In the plasma, the major finding was a depletion of mAPC levels with worsening hyperoxic lung injury. The importance of PAI-1 to the pathogenesis of hyperoxic lung injury is endorsed by the survival advantage of the PAI-1 knockout mouse, suggesting that the alveolar fibrin deposition in hyperoxia may be a significant contributor to lung injury and mortality.12 PAI-1, a significant inhibitor of fibrinolysis, when genetically deleted, may allow for enhanced clearance of intra-alveolar fibrin and subsequent improved gas exchange.
In this investigation mAPC plasma levels were significantly decreased when the lung was maximally injured. What is the mechanism of decreased mAPC plasma levels with hyperoxia? First, plasma levels of protein C, the substrate for mAPC production, remained unaltered throughout the course of hyperoxic lung injury. Second, TM and EPCR are the endothelial molecular machinery needed for mAPC production. While the plasma levels of TM and EPCR are unchanged with late hyperoxia, TM immunostaining reveals a severe loss of lung endothelial protein with moderate to severe hyperoxic lung injury. The late increase in soluble TM may reflect increased cleavage of TM during the severe endothelial injury phase of the hyperoxia model. Finally, plasma levels of PAI-1, a major inhibitor of mAPC, were unchanged with hyperoxia. These results suggest that the mechanism of mAPC depletion in hyperoxia is not the loss of substrate (protein C) or an increase in a dominant inhibitor of mAPC, but rather a defect in mAPC production from severe lung endothelial damage. This conclusion is further supported by the inability of thrombin challenge to restore mAPC plasma levels with severe hyperoxic lung injury. This pathological response to hyperoxia shares similarities to the response demonstrated in humans during meningococcaemia in which TM and EPCR immunostaining in skin endothelium is significantly decreased.20
What is the pathogenic significance of mAPC depletion in hyperoxia? It is well known that APC possesses several properties that could potentially be beneficial in hyperoxic lung injury. Acting through EPCR and PAR-1, APC has significant endothelial barrier protecting properties in vitro21 22 and improves mortality from experimental endotoxaemia and sepsis in vivo.23 The anti-apoptotic properties of APC24 25 could also be of benefit in hyperoxia, which is known to produce significant apoptosis, especially to the alveolar epithelium.26 APC also inhibits PAI-1 which, as previously discussed with the PAI-1 knockout studies, could be of benefit. Finally, human studies of pulmonary oedema fluid and plasma from patients with ALI/ARDS have revealed that protein C levels are decreased and PAI-1 levels are increased in the oedema fluid and plasma compartments, and mortality was higher with an increasing procoagulant antifibrinolytic profile.9 10 27 Therefore, from several lines of reasoning, decreased plasma levels of mAPC in hyperoxia could be detrimental.
We tested both preventive and treatment strategies with recombinant mAPC in hyperoxia, a model of lung injury for which many different prevention and treatment strategies have been effective.28–32 Other investigators have predominately used human APC in animal models of lung injury,19 33 although the specificity of human APC in these animals is unknown. We used the intratracheal route of administration as this was the only route tested that yielded sustained lung (BAL) and plasma levels of mAPC and has been used successfully by another group.19 However, intratracheal delivery of mAPC (1 mg/kg) in both treatment and preventive strategies did not ameliorate lung injury, despite having a biological effect on the procoagulant environment by reducing BAL fluid levels of PAI-1. Selected experiments with human APC were also ineffective. It has also recently been reported in a randomised placebo-controlled phase II clinical trial that APC was ineffective in the treatment of a cohort of patients with ALI but without severe sepsis.34
Our investigation has some limitations. We only measured one inhibitor of APC (ie, PAI-1). Other known inhibitors of APC include α1-antitrypsin, protein C inhibitor, and α2-macroglobulin, which could potentially increase with hyperoxia and compete with plasma mAPC. Second, given the time course of our model (several days) and mAPC delivery, we cannot conclude that mAPC replacement does not ameliorate non-infectious lung injury. However, we did deliver mAPC directly to the target organ in hyperoxia without any beneficial effect. Finally, it is possible that there is not only alveolar epithelial apoptosis with hyperoxia, but also a significant contribution of epithelial necrosis35 which could potentially limit the effectiveness of APC.
In conclusion, hyperoxia in mice produces significant damage and dysfunction to the lung endothelium and alveolar epithelium that is accompanied by local procoagulant changes and reduced plasma levels of mAPC. While the substrate for mAPC remains intact with hyperoxia, the dysfunctional endothelium limits mAPC generation. Through several potential mechanisms, mAPC depletion could be detrimental to the host in hyperoxic acute lung injury, although prevention or treatment strategies with mAPC did not ameliorate lung injury.
Acknowledgments
The authors acknowledge the technical assistance of Xiao Su, Jerrod Hunter and Mei Cheng and the statistical assistance of Hanjing Zhuo.
REFERENCES
Supplementary materials
web only appendix 64/2/114
Files in this Data Supplement:
Footnotes
See Editorial, p 95
▸ Additional data are published online only at http://thorax.bmj.com/content/vol64/issue2
Funding: This work was supported in part by a GlaxoSmithKline Pulmonary Fellowship and HL82742 (MRL), HL51854 and HL74005 (MAM), and HL54502 (CTE) from the National Institutes of Health.
Competing interests: MRL and MAM have no competing interests. CTE holds patents on production of protein C and on the use of activated protein C in sepsis. He also consults with several companies interested in sepsis and applications of anticoagulants to disease processes.