Article Text

Abstract
Background: Mast cell microlocalisation within the airway smooth muscle (ASM) bundle is an important determinant of the asthmatic phenotype. We hypothesised that mast cells migrate towards ASM in response to ASM derived chemokines.
Methods: Primary ASM cultures from subjects with and without asthma were stimulated with interleukin (IL)-1β, IL-4, and IL-13 alone and in combination. Mast cell chemotaxis towards these ASM supernatants was investigated, and the chemotaxins mediating migration by using specific blocking antibodies for stem cell factor (SCF) and the chemokine receptors CCR3, CXCR1, 3 and 4 as well as the Gi inhibitor pertussis toxin and the tyrosine kinase inhibitor genistein were defined. The concentrations of CCL11, CXCL8, CXCL10, TGF-β, and SCF in the supernatants were measured and the effect of non-asthmatic ASM supernatants on the mast cell chemotactic activity of asthmatic ASM was examined.
Results: Human lung mast cells and HMC-1 cells migrated towards Th2 stimulated ASM from asthmatics but not non-asthmatics. Mast cell migration was mediated through the combined activation of CCR3 and CXCR1. CCL11 and CXCL8 expression by ASM increased markedly after stimulation, but was similar in those with and without asthma. ASM supernatants from non-asthmatics inhibited mast cell migration towards the asthmatic ASM supernatant.
Conclusion: Th2 stimulated ASM from asthmatics is chemotactic for mast cells. Non-asthmatic ASM releases a mediator or mediators that inhibit mast cell migration towards stimulated asthmatic ASM. Specifically targeting mast cell migration into the ASM bundle may provide a novel treatment for asthma.
- ASM, airway smooth muscle
- FEV1, forced expiratory volume in 1 second
- HLMC, human lung mast cell
- IL, interleukin
- IFN-γ, interferon γ
- SCF, stem cell factor
- TGFβ, transforming growth factor β
- Th2, T helper 2
- mast cells
- chemokine receptors
- chemokines
- airway smooth muscle
- asthma
Statistics from Altmetric.com
- ASM, airway smooth muscle
- FEV1, forced expiratory volume in 1 second
- HLMC, human lung mast cell
- IL, interleukin
- IFN-γ, interferon γ
- SCF, stem cell factor
- TGFβ, transforming growth factor β
- Th2, T helper 2
Asthma is a common disease and remains a significant cause of morbidity and mortality worldwide.1 It is characterised by the presence of variable airflow obstruction, airway hyperresponsiveness, eosinophilic airway inflammation, T helper 2 (Th2) cytokine expression, and increased deposition of extracellular matrix beneath the basement membrane.2,3 We have recently shown that, in addition to these features, the airway smooth muscle (ASM) in asthma is infiltrated by mast cells. In contrast, mast cells within the ASM bundle was not a feature of non-asthmatic eosinophilic bronchitis or healthy controls, suggesting that this is a major determinant of the asthmatic phenotype.4–,6
It is likely that mast cells migrate towards the ASM bundle in asthma under the influence of ASM derived chemotaxins. Stem cell factor (SCF) is produced by human ASM and is chemotactic for mast cells,7,8 as well as being essential for their differentiation, maturation, proliferation and survival (reviewed by Page et al9). A role for SCF in the microlocalisation of mast cells to the ASM in asthma has therefore been proposed and needs further investigation. Alternatively, mast cell migration may be mediated by chemokines. We have recently shown that the chemokine receptors most commonly expressed by human lung mast cells are CCR3, CXCR1, CXCR3 and CXCR4.10 Supernatants derived from ASM from asthmatics stimulated with Th1 cytokines in vitro had greater mast cell chemotactic activity than ASM from normal controls.11 Interestingly, the ASM from these asthmatics preferentially released the CXCR3 ligand CXCL10 compared with ASM from non-asthmatics and CXCR3 blockade inhibited the mast cell chemotactic activity. The CXCL10/CXCR3 axis is therefore important in the migration of mast cells towards ASM in asthma. Although CXCL10 expression is upregulated in stable asthma12 and after allergen challenge,13–,15 there is a considerable body of evidence supporting the view that asthma is predominately a Th2 mediated disease.16
In asthma there is increased Th2 cytokine expression in bronchoalveolar lavage (BAL) fluid,17 sputum,18 and bronchial biopsy specimens17,19 in stable disease and after allergen challenge20 and, in addition, the mast cells in the ASM bundle in asthma express IL-4 and IL-13 on their surface.21 Thus, whether ASM stimulated with Th2 cytokines is also chemotactic for human mast cells needs to be examined. In this study we have tested our hypothesis that human lung mast cells (HLMC) and HMC-1 cells migrate in response to chemotaxins secreted by ASM from subjects with asthma, but not non-asthmatics, stimulated by Th2 cytokines.
METHODS
Subjects
Subjects were recruited from Sydney, Australia with two additional asthmatics recruited from Leicester, UK. Those with asthma gave an appropriate history and had objective evidence of variable airflow obstruction as indicated by one or more of the following: (1) airway hyperresponsiveness (methacholine concentration required to cause a 20% fall in forced expiratory volume in 1 second (PC20 FEV1) <8 mg/ml or maximum cumulative mannitol dose required to cause a 15% fall in FEV1 (PD15 FEV1) <635 mg/ml); (2) >15% improvement in FEV1 10 minutes after 200 μg inhaled salbutamol; or (3) peak expiratory flow (PEF) (>20% maximum within day amplitude from twice daily PEF measurements over 14 days). Subjects with asthma underwent bronchoscopy. Non-asthmatic subjects were undergoing lung resection for either lung transplantation or carcinoma. The study was approved by the Leicestershire and Sydney Central Area Health Service and University of Sydney Human Ethics Committees and all patients gave their written informed consent.
Airway smooth muscle isolation and culture
From bronchial biopsy specimens or resected bronchial tissue, pure ASM bundles were dissected free of surrounding tissue. In the non-asthmatics the ASM was derived from similar airway divisions to the bronchial biopsies. The small muscle bundles were cultured in DMEM supplemented with 10% FBS, 4 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin. ASM cell characteristics were determined by immunofluorescence and light microscopy with α-smooth muscle actin-FITC direct conjugate and calponin and myosin indirectly conjugated with FITC (Sigma, Gillingham, Dorset, UK).
Mast cell isolation and culture
HLMC from normal lung obtained at surgery for carcinoma at Leicester, UK (seven donors) were isolated by immunomagnetic affinity purification using the Dynal Cellection kit (Dynal, Oslo, Norway) as previously described.22. A typical lung sample provided a yield of 0.1−0.5×106 HLMC/g of lung of >98% purity. Cells were cultured in DMEM, 10% fetal bovine serum (FBS) supplemented with SCF (100 ng/ml), IL-10 (10 ng/ml), and IL-6 (50 ng/ml) (R&D Systems, Abingdon, Oxfordshire, UK).
HMC-1 cells
The HMC-1 cell line was a gift from Dr J Butterfield (Mayo Clinic, Rochester, MN, USA). HMC-1 cells were maintained in Iscove’s modified DMEM as described previously.23
ASM stimulation
ASM cells from nine asthmatic and six non-asthmatic subjects were plated into 6-well plates (9.6×104 cells in 2 ml DMEM and 10% FBS), grown for 1 week, then growth was arrested for 48 hours with serum deprived medium and either left unstimulated or stimulated with IL-1β and/or Th2 cytokines as follows: (i) IL-1β, (ii) IL-4, (iii) IL-13, (iv) IL-4 and IL-13, and (v) IL-1β, IL-4 and IL-13 (10 ng/ml of each; R&D Systems, Abingdon, UK) for 24 hours. Using the same protocol, further ASM from five asthmatics and six non-asthmatics were stimulated with Th1 cytokines IL-1β, TNFα and IFN-γ (10 ng/ml each cytokine in combination; R&D Systems). The cytokine concentrations were chosen to reflect the maximal response as identified in previous reports.24 Supernatants were collected and stored at −80°C and cell counts were derived for the cells in each well. Experiments were performed using ASM cells from passage 3–7 and the passage number was not different between ASM from subjects with or without asthma.
Insufficient supernatants were available to study the role of SCF in mast cell migration to Th2 stimulated ASM supernatants. Thus, additional ASM supernatants from seven asthmatics and eight non-asthmatics where non-confluent cells had been left unstimulated or stimulated with IL-4 and IL-13 (20 ng/ml) for 48 hours after serum deprivation for 24 hours were used.
Chemotaxis assays
Chemotaxis assays were performed using Transwells with fibronectin coated inserts with a pore size of 8 μm (BD, Oxford, UK) as previously described.10 We placed 1×105 HLMC or 2×105 HMC-1 cells in 100 μl culture medium supplemented with 2% FCS into the top well, and 450 μl of supernatant from stimulated ASM or appropriate negative control in the bottom well. Checkerboard analysis was used to distinguish chemotactic from chemokinetic activity. Mast cells were preincubated with pertussis toxin (0.5 μg/ml) for 1 hour and 16 hours and with the tyrosine kinase inhibitor genistein (0.5 μg/ml) for 1 hour before the chemotaxis assays to assess the effect of these inhibitors on migration as previously described.24 Similarly, to assess the relative importance of individual chemokine receptors, we preincubated the cells with receptor blocking antibodies CCR3, CXCR1, CXCR3 and CXCR4 (R&D Systems) alone or in combination or isotype controls for 1 hour. After preincubation with inhibitors, the cells were washed and recounted to ensure that the number of cells added to the top chamber of the chemotaxis wells was consistent across conditions. The role of SCF was examined using the chemotaxis assay outlined above with ASM supernatants or recombinant SCF (50 ng/ml) in the bottom well preincubated with SCF neutralising antibody or an isotype control for 0.5 hours (R&D Systems).
We considered that ASM supernatants from non-asthmatics might affect the chemotactic activity of asthmatic ASM supernatants. To investigate this possibility we assessed mast cell chemotaxis towards Th1 and Th2 stimulated ASM supernatants from asthmatics after titration with media alone or with the corresponding Th1 or Th2 stimulated non-asthmatic ASM supernatants using the chemotaxis assay described above. In these chemotaxis assays the final concentration of the asthmatic ASM supernatant as a proportion of neat supernatant was 12.5–75%.
Concentration of mediators in conditioned ASM supernatants
The concentrations of several putative mast cell chemotaxins were measured in the Th2 stimulated ASM supernatants by commercial ELISA: CCL11 (BD), CXCL8 (BD), CXCL10 (BD), SCF (R&D), and TGF-β (R&D). In addition, we measured using ELISA, in Th1 and Th2 stimulated ASM supernatants, PGE2 (Cayman Chemical, Ann Arbor, MI, USA) and IFN-β (R&D) as we considered that these mediators may play a role in modulating the migration of mast cells towards ASM supernatants. The concentration of mediators was corrected for cell number. The limits of detection were as follows: CCL11 (6.25 pg/ml), CXCL8 (3.1 pg/ml), CXCL10 (31.5 pg/ml), SCF (15.63 pg/ml), TGF-β (31.25 pg/ml), PGE2 (15 pg/ml), and IFN-β (250 pg/ml).
Statistical analysis
HLMC and HMC-1 migration was expressed as the geometric mean (SE) fold difference in migration compared with control and the effect of blocking antibodies and inhibitors was expressed as mean (SE) % inhibition. Chemokine concentrations for CCL11 and CXCL8 were log normally distributed and were described as the geometric mean (logSE); the other chemotaxins or mediators were described as median (interquartile range). The mast cell chemotactic index for ASM stimulated with each condition was assessed by t test. Comparison between mast cell migration towards asthmatic versus non-asthmatic ASM stimulated with different conditions were analysed by ANOVA across conditions and by unpaired t tests for each condition separately. A value of p<0.05 was taken as statistically significant.
RESULTS
Chemotactic activity for HMC-1 and HLMC by asthmatic ASM
Supernatants of ASM cells from asthmatic subjects (n = 7) were markedly more chemotactic for HMC-1 cells than those from non-asthmatic subjects (n = 5) when the ASM cells were activated with IL-1β or IL-13 alone, IL-4 and IL-13 in combination, or with IL-1β, IL-4 and IL-13 in combination (fig 1A⇓). The cell number from the asthmatic ASM cultures (3.2 (0.4) ×105cells/well) was increased compared with the non-asthmatic ASM cultures (1.1 (0.3) × 105cells/well; p = 0.001), so migration assays were performed towards the IL-1β, IL-4 and IL-13 stimulated ASM cell supernatants with the supernatant diluted to correct for the difference in cell number. Even after this correction the supernatants from the asthmatic ASM remained chemotactic for mast cells (2.2-fold compared with control media; p = 0.008) and was increased compared with those from non-asthmatics (1.3-fold v 2.2-fold; p = 0.025; fig 1B⇓). HMC-1 migration towards ASM stimulated with IL-1β, IL-4 and IL-13 in combination (n = 4) was not inhibited by CCR3, CXCR1, CXCR3 or CXCR4 blocking antibodies alone but was in combination (94 (6)% inhibition compared with isotype control; p<0.001). Isotype controls did not affect HMC-1 migration (data not shown). HMC-1 migration towards triple stimulated ASM was also inhibited by genistein and pertussis toxin (fig 2A⇓).
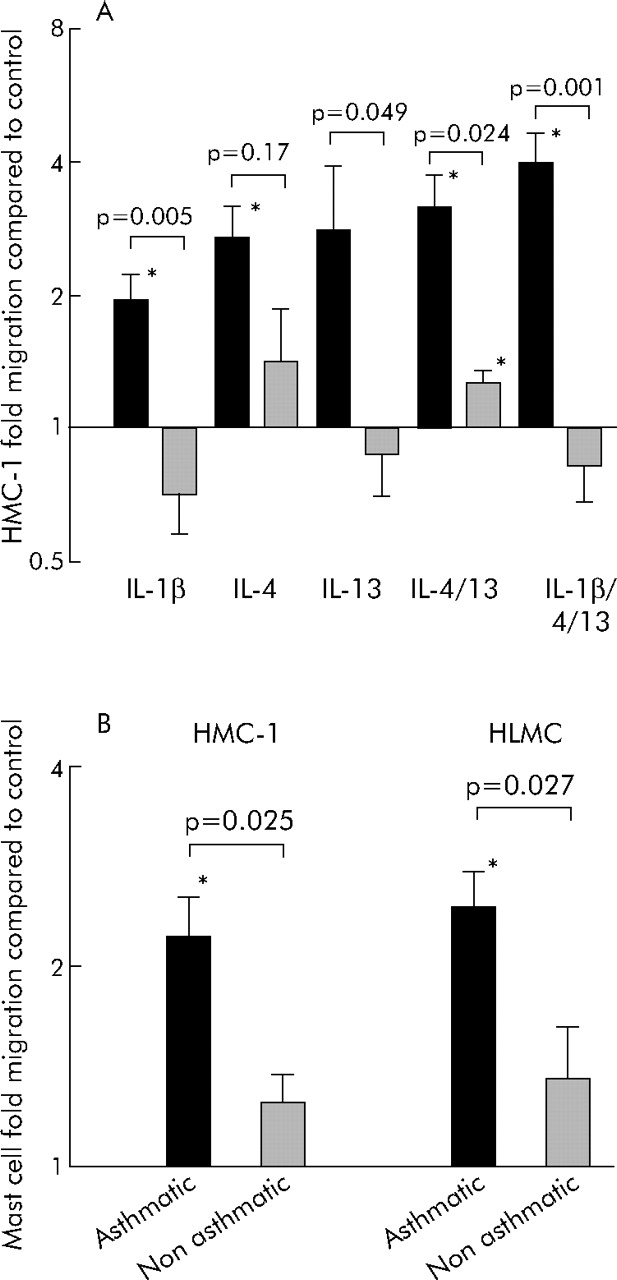
(A) Mean (SE) HMC-1 migration towards asthmatic and non-asthmatic ASM stimulated with IL-1β, IL-4 and IL-13 alone or in combination. Solid bars represent asthmatic ASM, hatched bars respresent non-asthmatic ASM. (B) Mean (SE) HMC-1 and HLMC migration towards asthmatic and non-asthmatic ASM stimulated with IL-1β, IL-4 and IL-13 in combination after supernatants were corrected for cell number. Stated p values are for comparisons of mast cell migration towards asthmatic v non-asthmatic ASM; *p<0.05 for mast cell migration towards ASM conditioned media compared with control media. p values on the figure represent comparisons between asthmatics and non-asthmatics.

Mean (SE) % inhibition of (A) HMC-1 and (B) HLMC migration towards asthmatic ASM stimulated with IL-1β, IL-4 and IL-13 in combination after preincubation of mast cells with chemokine receptor blocking antibodies, pertussis toxin (PTx), or genistein (*p<0.05).
Similarly, IL-1β, IL-4 and IL-13 stimulated ASM supernatants from asthmatics (n = 6), after correction for cell number, were chemotactic for HLMC (2.4-fold v control media; p = 0.002), but not ASM supernatant from non-asthmatics (n = 7; 1.4-fold; p = 0.2). There was a significant difference between HLMC migration towards asthmatic compared with non-asthmatic ASM supernatant (p = 0.03; fig 1B⇑). HLMC migration towards asthmatic ASM stimulated with IL-1β, IL-4 and IL-13 in combination (n = 4) was inhibited by pertussis toxin; CCR3, CXCR1, CXCR3 and CXCR4 blocking antibodies in combination (51 (11)% inhibition; p = 0.043); and CCR3 and CXCR1 blocking antibodies in combination (59 (5)% inhibition; p = 0.006) compared with media alone with or without isotype control, but not genistein (fig 2B⇑).
HMC-1 chemotaxis to the asthmatic and non-asthmatic ASM supernatants was not affected by SCF neutralising antibody (data not shown), although HMC-1 chemotaxis to SCF (50 ng/ml) was inhibited by 97.6 (0.9)% (p<0.001, n = 3).
Checkerboard analysis confirmed that HMC-1 and HLMC migration towards Th2 stimulated asthmatic ASM supernatants was due to chemotaxis rather than chemokinesis. IL-1β, IL-4 and IL-13 in the medium at concentrations used to stimulate the ASM did not induce HMC-1 or HLMC migration (data not shown).
Chemokine expression by ASM
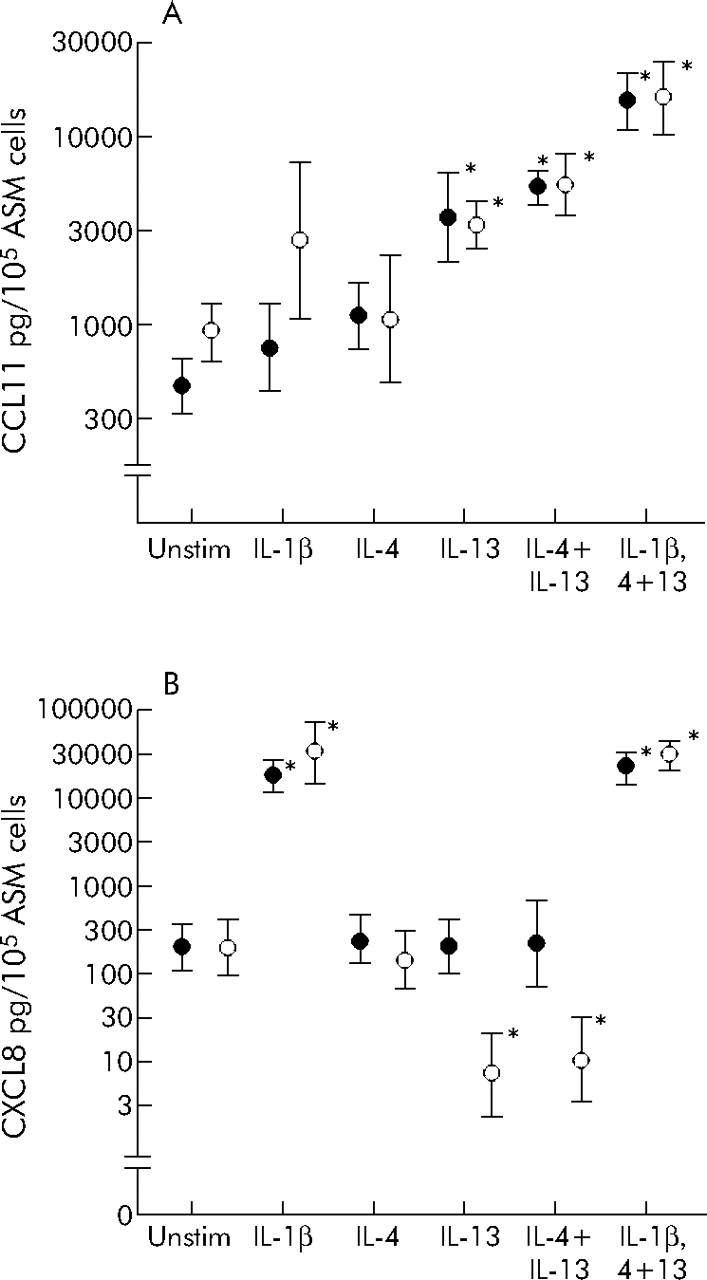
The concentration of CCL11 was increased in the ASM supernatants after stimulation with IL-13 alone or in combination with IL-4 and IL-1β (fig 3A⇓). However, there was no difference in CCL11 concentration between ASM from asthmatic (n = 7) or non-asthmatic subjects (n = 5) before or after stimulation once the supernatants were corrected for cell number. CXCL8 concentration was increased in the supernatants stimulated with IL-1β alone or in combination with IL-4 and IL-13. The CXCL8 concentration did not increase with IL-4 or IL-13 stimulation alone or in combination. Indeed, the CXCL8 concentration decreased after stimulation with IL-13 alone or in combination with IL-4 in the non-asthmatic but not in the asthmatic ASM supernatants (fig 3B⇓).

Geometric mean (SE) concentration of (A) CCL11 and (B) CXCL8 in the supernatants from stimulated ASM from asthmatics (closed symbols) and non-asthmatics (open symbols). *p<0.05.
In the ASM supernatants stimulated with IL-1β, IL-4 and IL-13, the concentrations of CXCL10 and TGFβ were low and not increased in those from asthmatic (52 (16–198) and 20 (2–27) pg/105 cells) compared with non-asthmatic subjects (258 (72–506) and 36 (0–84) pg/105 cells). Likewise, similar amounts of soluble SCF were detected in the unstimulated supernatants of ASM cells from seven asthmatic subjects (87.6 (63–151) pg/105 cells) and eight non-asthmatic subjects (128 (84–136) pg/105 cells), and stimulation with the Th2 cytokines IL-4 and IL-13 did not alter their SCF release (data not shown).
Asthmatic ASM chemotactic activity attenuated by non-asthmatic ASM
Supernatants of ASM cells from three asthmatic subjects stimulated with Th1 (IL-1β, TNFα and IFN-γ) or Th2 (IL-1β, IL-4 and IL-13) cytokines were markedly more chemotactic for HMC-1 than those from three non-asthmatic subjects (fig 4⇓). This chemotactic activity of asthmatic ASM was attenuated by titration with media alone, but inhibition of chemotaxis was more marked by titration with non-asthmatic ASM supernatants (fig 4⇓).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean (SE) HMC-1 migration towards neat (A) Th1 stimulated and (B) IL-1β, IL-4 and IL-13 stimulated ASM supernatants from three asthmatic and three non-asthmatic donors, and towards stimulated asthmatic ASM supernatants after titration with media alone or with non-asthmatic stimulated ASM supernatants. Final concentration of asthmatic ASM supernatant was 12.5–75% of neat supernatant. *p<0.05 HMC-1 migration towards supernatant compared with control.
PGE2 concentrations in ASM supernatants stimulated with IL-1β, IL-4 and IL-13 (Th2 stimulated) or IL-1β, TNFα and IFN-γ (Th1 stimulated) were not significantly different in subjects with asthma (1 (4–37) ng/105cells and 17 (13–22) ng/105cells) and non-asthmatic subjects (3 (2–30) ng/105cells and 17 (15–20) ng/105cells respectively; p>0.05). IFN-β was undetectable in all of the Th1 and Th2 stimulated samples.
DISCUSSION
We have made several new and important observations. Supernatants from Th2 stimulated ASM from subjects with asthma, but not non-asthmatics, induced HLMC and HMC-1 migration. This was mediated predominantly through activation of CCR3 and CXCR1, even though ligands for these receptors, CCL11 and CXCL8 were not released preferentially by asthmatic ex vivo ASM cells compared with those from non-asthmatic subjects. Importantly, ASM supernatants from non-asthmatics inhibited the mast cell chemotactic activity of Th1 and Th2 stimulated asthmatic ASM. These observations suggest that ASM from non-asthmatics release an inhibitory factor or factors that attenuate chemokine mediated migration of mast cells towards ASM.
In asthma the ASM bundle is infiltrated by mast cells.4 This is not a feature of eosinophilic bronchitis or normal controls, suggesting that mast cell-ASM interactions may be fundamental in the development of the asthma phenotype. It is therefore likely that mast cells migrate to ASM under the influence of chemotaxins released by ASM. Indeed, we found that supernatants from Th2 stimulated ASM from asthmatics, but not non-asthmatic subjects, were chemotactic for HLMC and HMC-1 cells. Similarly, we confirmed that Th1 stimulated asthmatic ASM had increased mast cell chemotactic activity compared with non-asthmatic ASM.11 The lack of chemotactic activity in the Th2 stimulated ASM supernatants from the non-asthmatic subjects is consistent with the paucity of mast cells in the healthy ASM in bronchial biopsies. Blocking the chemokine receptors most commonly expressed by HLMC (CCR3, CXCR1, CXCR3 and CXCR4) alone had little effect on mast cell migration towards the Th2 stimulated asthmatic ASM but, importantly, migration was inhibited markedly by Gi inhibitor pertussis toxin and by blocking CCR3 in combination with CXCR1. HMC-1 migration, but not HLMC migration, was inhibited partly by genistein, a tyrosine kinase inhibitor, which suggests that chemotaxins that activate this pathway such as TGF-β are unlikely to play a significant role in mast cell migration towards Th2 stimulated ASM supernatants. Taken together, our findings therefore suggest that activation of CCR3 and CXCR1 is the dominant mechanism mediating mast cell chemotaxis towards Th2 stimulated asthmatic ASM.
In this study we found, as reported by Hirst et al,25 that ASM released increased concentrations of the CCR3 ligand CCL11 after Th2 stimulation and the CXCR1 ligand CXCL8 after stimulation with IL-1β. Intriguingly, the concentrations of these chemokines were not different between asthmatic and non-asthmatic subjects. The concentrations of these chemokines in the ASM supernatants cannot therefore explain the differential migration towards Th2 stimulated asthmatic and non-asthmatic ASM supernatant. One possible explanation for this apparent anomaly is that other CCR3 and/or CXCR1 chemokines are released preferentially by asthmatic ASM. However, recombinant CCL11 and CXCL8 at similar concentrations to those released by Th2 stimulated non-asthmatic ASM are chemotactic for mast cells.10,26,27 Therefore, an alternative explanation for the different mast cell chemotactic activity between asthmatic and non-asthmatic ASM is that a mediator released by non-asthmatic ASM inhibits mast cell migration. In support of this view we found that the mast cell chemotactic activity of Th2 stimulated ASM was inhibited by titration with non-asthmatic ASM more markedly than titration with media alone. The inhibitory capacity of non-asthmatic ASM supernatant was not reserved to ASM stimulated with Th2 cytokines. Indeed, we found that mast cell migration towards Th1 stimulated asthmatic ASM was likewise inhibited by non-asthmatic ASM. We have considered a number of possible explanations for this phenomenon. The CXCR3 ligands are natural antagonists for CCR328 and therefore have the capacity to inhibit CCR3 mediated migration in the Th2 stimulated asthmatic ASM. However, the CXCL10 concentration was low and was not increased in the Th2 stimulated asthmatic ASM, so it is unlikely to have influenced mast cell migration. PGE2 secretion by asthmatic ASM in response to serum is reduced,29 but we found it was not different between asthmatics and non-asthmatics after Th1 or Th2 stimulation, in keeping with earlier findings.30 This suggests that this mediator is unlikely to be responsible for the differential mast cell migration towards asthmatic and non-asthmatic ASM. IFN-β has been implicated in modifying chemokine release by ASM via an autocrine mechanism,31 but we were unable to detect IFN-β in samples from asthmatics or non-asthmatics stimulated with Th1 or Th2 cytokines, suggesting that this mediator is also unlikely to affect mast cell migration towards ASM supernatants. In addition, the non-asthmatic ASM was derived from airways of a similar division to bronchial biopsies and the ASM passage number did not differ between asthmatic and non-asthmatic subjects, which suggests that these technical issues cannot explain our findings. The mechanism by which non-asthmatic ASM inhibits mast cell migration therefore remains unexplained and needs to be further examined.
In this study mast cell migration towards Th2 stimulated asthmatic ASM was mediated by activation of CCR3 and CXCR1 in combination. However, it is likely that mast cell migration towards the ASM in asthma is under the control of several chemotaxins, and the relative importance of each of these varies depending upon the inflammatory environment within the individual asthmatic. In an earlier study we found that mast cell migration towards the ASM bundle in asthma was mediated via CXCL10 activation of CXCR3.11 ASM bundles in bronchial biopsies from asthmatics expressed CXCL10 constitutively and, following Th1 stimulation, asthmatic ASM preferentially released CXCL10. ASM activated by tryptase induced mast cell chemotaxis after the production of TGF-β1 and SCF.7 Th2 stimulation did not increase CXCL10, TGF-β and SCF expression and SCF was not involved in ASM induced mast cell chemotaxis, supporting the view that the type of inflammatory stimulus determines the dominant mast cell chemotaxin released by ASM. Further work is required to identify the important factors that may be involved in mast cell migration towards ASM stimulated by mast cells themselves. It also remains a possibility that it is mast cell precursors that are recruited by the ASM rather than mature airway cells. Interestingly, the expression of CCR3 on human bone marrow derived mast cells is similar to mature lung mast cells,10 suggesting that this chemokine receptor may be particularly important in the recruitment of progenitors to tissue.32
In summary, our findings show that ASM from asthmatics, when stimulated with Th2 cytokines, is chemotactic for mast cells predominantly mediated by activation of mast cell CCR3 and CXCR1. Non-asthmatic ASM releases a mediator or mediators that inhibit mast cell migration towards both Th1 and Th2 stimulated asthmatic ASM. Specifically targeting mast cell migration into ASM bundles may provide a novel effective treatment for asthma.
Acknowledgments
The authors thank the theatre and pathology staff of the Sydney metropolitan hospitals and University Hospitals of Leicester for the supply of human lung tissue and the collaborative effort of the cardiopulmonary transplant team at St Vincent’s Hospital, Sydney. They also thank Mrs D Parker for technical support.
REFERENCES
Footnotes
Published Online First 6 April 2006
Supported by Asthma UK, DoH UK Clinician Scientist Scheme, NHMRC Australia.
Competing interests: none.