Article Text

Abstract
Background: Cystic fibrosis (CF) airways are characterised by chronic inflammation, increased interleukin (IL)-8 secretion, and neutrophil activation which are considered the principal factors of morbidity and mortality in CF patients. Optimising management of this chronic inflammatory response is therefore a key issue of basic and clinical CF research. Several reports have addressed ways to manage CF airways inflammation, and an attractive therapeutic strategy may be the inhibition of the p38-mitogen activated protein kinase (p38-MAP-k) pathway.
Methods: A new ex vivo model was used to study the mucosal inflammatory response to environmental airways stimuli. Nasal biopsy tissues from CF patients and controls were cultured ex vivo for 20 minutes, 4 hours, and 24 hours in the presence of lipopolysaccharide (LPS) from Pseudomonas aeruginosa (PA) with and without the p38-MAP-k inhibitor SB203580. Quantitative mRNA assessment, immunohistochemistry, and Western blots were used to detect the expression and modulation of inflammatory markers.
Results: PA-LPS challenge induced a time dependent mucosal inflammation indicated by rapid epithelial activation, IL-8 release, COX-2 upregulation, and neutrophil migration to the upper mucosal layers. Some of these LPS induced changes (IL-8 release and neutrophil migration) were specific to CF tissues. SB203580 significantly controlled all LPS induced mucosal changes in CF tissues.
Conclusion: These findings provide a rationale and proof of principle for the potential use of p38-MAP-k inhibitors to control inflammation in patients with CF.
- CF, cystic fibrosis
- IL, interleukin
- LPS, lipopolysaccharide
- MAP-k, mitogen activated protein kinase
- PA, Pseudomonas aeruginosa
- interleukin 8
- neutrophil
- inflammation
- cystic fibrosis
- mitogen activated protein kinase
Statistics from Altmetric.com
- CF, cystic fibrosis
- IL, interleukin
- LPS, lipopolysaccharide
- MAP-k, mitogen activated protein kinase
- PA, Pseudomonas aeruginosa
Cystic fibrosis (CF), caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, is one of the most common inherited lethal diseases with an incidence of the order of 1/2500.1 It is a multisystemic disease with a complex clinical pattern as CFTR is expressed in subsets of epithelial cell types. Chronic respiratory infections in CF, initially dominated by Staphylococcus aureus and subsequently by Pseudomonas aeruginosa (PA), force persistent inflammation and progressive lung damage which is the main cause of morbidity and ultimately mortality in patients with CF.1 These chronic infections are therefore considered to be key factors in sustaining inflammation which might find a fertile ground in the “intrinsic” aberrant inflammatory response characteristic of CF patients,1–3 apparently related to the activation of the NF-κB pathway,2,4–6 but a deficit of the lipoxin pathway might also be involved.7 It has to be stressed, however, that the theory of intrinsically dysregulated inflammation is controversial.8 Despite the presence of chronic infections, the infiltrate of patients with CF is characterised by the absence of lymphocyte responses. All these aspects make the inflammatory response in CF atypical. Clinical trials of gene therapy in CF, which is considered to be the prototype of the diseases treatable in this way, have not provided the expected results.9 In the absence of definitive therapy, these patients are treated with antibiotics1 as well as anti-inflammatory regimens to reduce inflammation.1,10 A series of anti-inflammatory drugs such as ibuprofen, corticosteroids1,10 or, more recently, long term azithromycin11 have been used to treat patients with CF, without a satisfactory proof of principle for their effectiveness.
The lung of CF patients is characterised by several inflammatory alterations arising from an imbalance in pro-inflammatory signal transduction pathways. The main features of inflamed CF lungs are increased interleukin 8 (IL-8) production and neutrophil infiltration in the parenchyma with influx into the air space.1,2 It is therefore reasonable to propose that anti-inflammatory treatment of CF should target IL-8 production, thus suppressing neutrophil migration and activation.
We describe a new approach to test potential anti-inflammatory strategies in CF using an ex vivo organ tissue culture model which we have used in the study of other inflammatory conditions and potential strategies to modulate the inflammatory response.12,13 This model represents a good approximation to in vivo studies since, in cultured biopsy tissues, all the anatomical connections are retained and all cell types (epithelial, myeloid, lymphoid) interact with neighbouring cells within their natural environment. For this study we have used explants of nasal polyps which sometimes complicate CF as they provide a suitable representation of CF chronic inflammation and are routinely removed surgically.14
Using this approach we have explored whether p38-mitogen activated protein kinase (p38-MAP-k) inhibition represents a suitable and attractive strategy for treating patients with CF. We targeted the p38-MAP-k pathway because of its role in IL-8 production in CF,15,16 its reported activation in CF cells17 and in vitro, as well as in vivo, neutralisation of neutrophil function.18 For this purpose the nasal polyp explants were stimulated with PA-LPS in the presence or absence of a specific and most widely used p38-MAP-k pathway inhibitor. Our study aims to test whether the inhibition of the p38-MAP-k pathway is effective in blocking IL-8 release, neutralising neutrophil migration and degranulation and other signs of CF inflammation. We provide evidence of proof of principle for the potential clinical use of p38-MAP-k inhibitors to control inflammation in CF.
METHODS
Patients and ex vivo cultures
Seven consecutive CF patients with chronic sinusitis and nasal polyposis and seven consecutive non-CF patients (mean age 21 years, range 16–32) with idiopathic polyposis underwent surgical treatment. All CF and control patients presented with a history of more than 2 years of chronic sinusitis with nasal obstruction; all had undergone a sinus computed tomographic scan during their evaluation. Surgery was used only when medical treatment had failed. All patients in both the CF and non-CF groups suspended the use of corticosteroids at least 1 month before the surgical intervention and were only treated by saline nasal irrigation. No patient had allergic rhinitis, as documented by clinical history. The patients were screened for reactivity to the most common inhaled allergens by prick tests and none tested positive.
Nasal polyp explants were cut into at least 10 small specimens within a few minutes after the surgical excision. One fragment from each patient was embedded in optimal cutting temperature (OCT) compound (Tissue Tek, Miles Laboratories, Elkhart, IN, USA) and stored at −80°C. The other fragments were cultured as previously reported for duodenal biopsies13,19 for 20 minutes, 4 hours, or 24 hours in the presence of medium alone, medium with 1 μg/ml lipopolysaccharide (LPS) from PA (PA-LPS; Sigma, St Louis, MO, USA), or PA-LPS with selective p38-MAP-k inhibitor SB20358020 (Calbiochem, San Diego, CA, USA) and stored at −80°C. SB203580 was used at a concentration of 1 μM considered to be specific for the p38-MAP-k, although effects on other signal transduction pathways might still occur.20 Since SB203580 was diluted 1/1000 from a 1 mM stock solution of DMSO, in all experiments DMSO was used at 1/1000 dilution as an internal control and the samples were analysed together with those cultured in the presence of medium, PA-LPS, or PA-LPS + SB203580. No effects of DMSO were observed in any patient. Culture supernatants were collected and stored at −80°C. Additional details of the ex vivo tissue culture model are provided in an online supplement available on the Thorax website at http://www.thoraxjnl.com/supplemental.
Fully informed consent was obtained from all subjects and the ethical committee of Regione Campania Health Authority approved the study.
COX-2 and IL-8 mRNA detection in tissue samples
Quantitative PCR assessment of COX-2 and IL-8 mRNA were performed as previously described.21 Additional details on the method are provided in the online supplement.
Cytokine detection in culture supernatants
Culture supernatants were analysed for the presence of IL-8, IL-1β, IL-6, IL-10, TNF-α and IL-12p70 at 4 hours by Cytometric Bead Array (BD-Biosciences, San Jose, CA, USA) using a FACScan (BD-Biosciences).
Antibodies and antigen detection in tissue samples
Five μm thick cryostat sections were incubated for 2 hours at room temperature with monoclonal antibodies against COX-2, NF-κB (p65 subunit), phosphotyrosine PY99, phospho-p38-MAP-k, cathepsin G, cytokeratin (5/6/8/18). Antigen expression was visualised by immunofluorescence or peroxidase staining. Details on antibodies and antigen detection are shown in table E1 in the online supplement.
COX-2 positive mononuclear cells (MNC) were counted per mm2 of mucosa. The distribution of neutrophils (cathepsin G positive polymorphonuclear (PMN) leucocytes) within the different mucosal compartments was analysed within standard reference areas. Epithelial cells with nuclear p65 localisation or with PY-99 or phospho-p38 expression were counted per 100 epithelial cells. Data were blindly evaluated by two independent observers unaware of the culture conditions. Additional details on the method are provided in the online supplement.
Neutrophil migration assay
The MIGRATEST Orpegen (Heidelberg, Germany) was used to study the effects of p38-MAP-k inhibition on in vitro neutrophil migration. The assay was performed according to the manufacturer’s specifications. Samples were analysed by flow cytometry using FACS (Beckman Coulter) with an argon laser tuned at 488 nm. The data were evaluated using WinMDI software.
Western blot analysis
Rabbit polyclonal anti-MAPKAP-k-2[pT334] (Biosource, Camarillo, CA) was used to dectect changes in p38-MAP-k activity. Further details are provided in the online supplement.
Statistical analysis
Paired data analysis was used to compare samples cultured with medium with those cultured with LPS or LPS + SB203580 in each set of experiments (20 min, 4 hours, and 24 hours). Before analysis, data were screened for gross deviance from normality. Since some groups of variables showed a skewness of >1 and the sample size was small, non-parametric tests (Wilcoxon’s test) for paired samples were applied throughout the study for the analysis of the number of the cells that expressed the tested markers. Non-parametric tests for unpaired samples (Mann-Whitney test) were used to compare CF and control samples for the expression of the tested markers before ex vivo challenge. Statistical analysis was performed using SPSS Version 11.1.
RESULTS
The clinical characteristics of the patients with CF are summarised in table 1. The control group was tested for the presence of the most commonly found mutations in the Mediterranean area and found to be negative.22
Summary of clinical characteristics of patients with CF
Effect of PA-LPS challenge on p65 nuclear localisation in airway epithelium
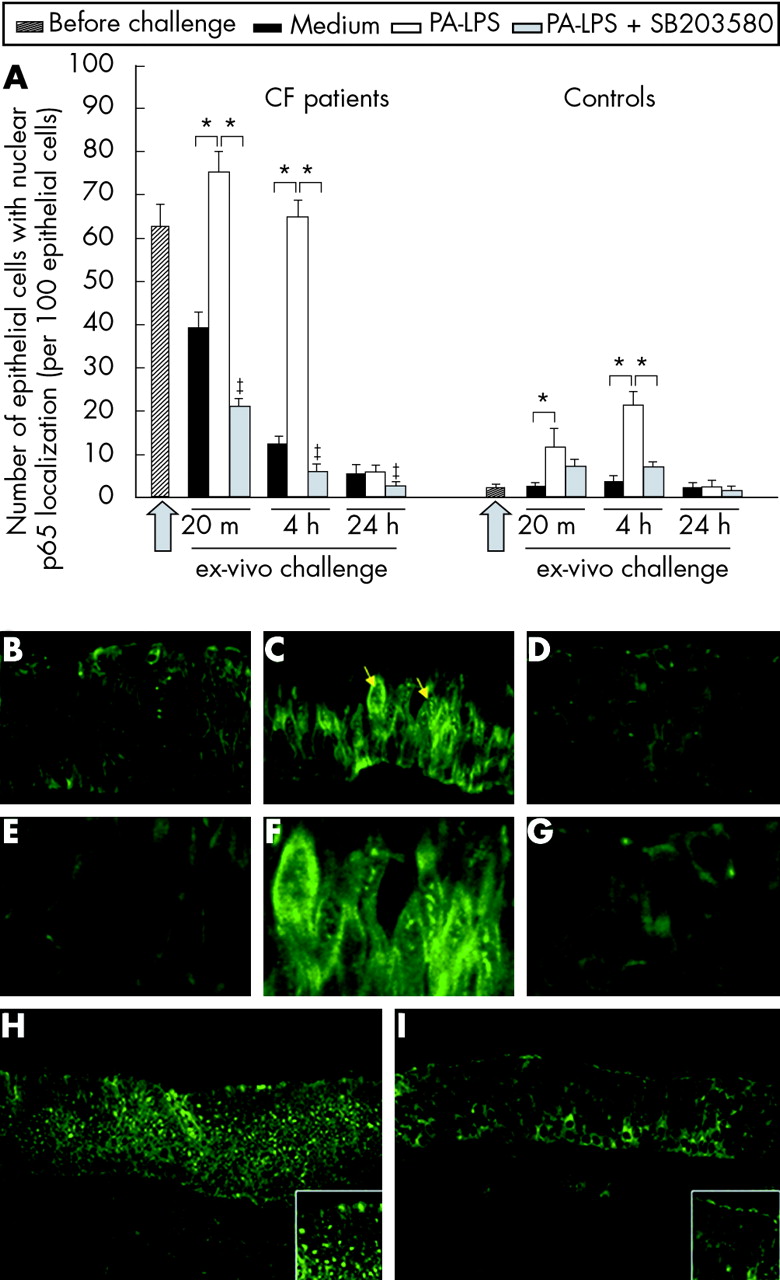
Higher numbers of epithelial cells with p65 nuclear localisation were observed in CF nasal polyps before challenge than controls (p = 0.002, fig 1A). PA-LPS challenge led to an increase in the number of airways epithelial cells with p65 nuclear localisation at 20 minutes and 4 hours of incubation (p = 0.018 v cultures with medium alone; fig 1A, B, C, E, F, H and fig E1 online only) which was not detected after 24 hours (fig 1A). This effect of PA-LPS was observed both in CF patients and controls (fig 1A). p65 nuclear localisation was significantly inhibited by SB203580 treatment (p = 0.018 v cultures with PA-LPS alone; fig 1A, D, G, and I); SB203580 also reduced the residual p65 localisation observed in CF biopsies cultured with medium alone (p = 0.018). The most obvious changes following PA-LPS stimulation were seen in epithelial cells. These data indicate that the in vitro culture model allows survival of the nasal explants. They also show that, in the absence of stimuli, NF-κB activation decreases in CF tissues (fig 1A) and they demonstrate the effectiveness of SB203580 treatment in controlling PA-LPS induced p65 nuclear localisation (fig 1A). Importantly, no significant structural differences were observed between CF and control tissues as described in fig E2 (online only) when stained with pancytokeratin antibody.
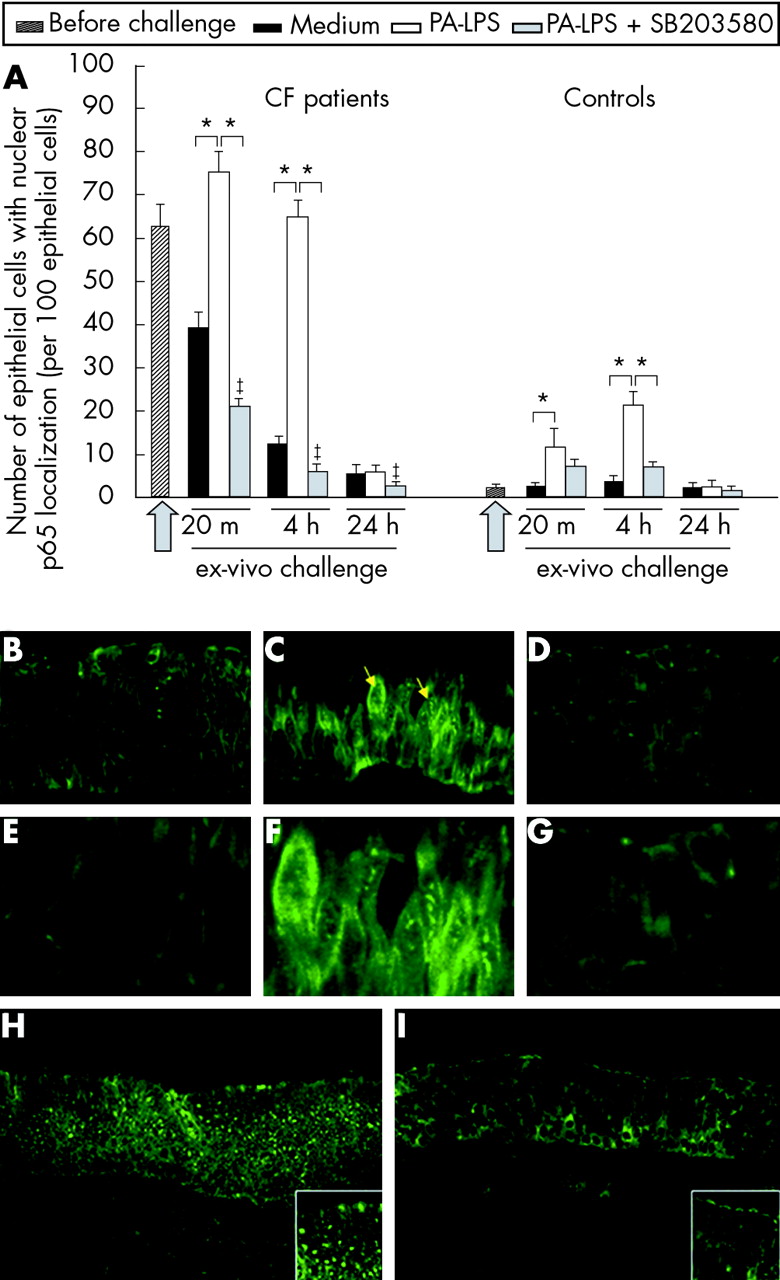
p65 nuclear localisation in the airway epithelium following PA-LPS challenge of nasal polyp explants from CF patients: effects of p38-MAP-k inhibition by SB203580. (A) Time course of PA-LPS induced p65 nuclear localisation in nasal polyp epithelial cells in CF and control tissues and effect of p38-MAP-k inhibition by SB203580. Mean (SD) number of epithelial cells with p65 nuclear localisation per 100 epithelial cells before challenge and after challenge with medium alone, PA-LPS, or PA-LPS + SB203580 following 20 minutes, 4 hours and 24 hours of incubation. The arrow indicates the start point of the challenge. *p<0.05 v samples cultured with medium alone and v samples cultured with PA-LPS + SB203580; ‡p<0.05 v samples cultured with medium alone (Wilcoxon’s test). Combined data obtained from all seven CF or all seven control samples. (B–G) Epithelial expression and localisation of p65 in CF nasal polyp explants after 4 hours of challenge in the presence of (B) medium alone, (C) PA-LPS, and (D) PA-LPS + SB203580; (E), (F) and (G) are higher magnifications of (B), (C) and (D), respectively. Faint p65 nuclear localisation is detected in a few epithelial cells after incubation with medium alone (B); the challenge with PA-LPS induces an increase in nuclear p65 localisation in the majority of epithelial cells (C, yellow arrows). Treatment with SB203580 prevented p65 nuclear localisation induced by PA-LPS challenge and almost all nuclei in the field appear devoid of any label (D). (H) and (I) Low magnification pictures showing a wide representative area of CF tissues after 4 hours of stimulation with (H) PA-LPS or (I) PA-LPS + SB203580. Note that the majority of epithelial cells show p65 nuclear localisation on PA-LPS stimulation, as detailed in the high magnification insert at the bottom right corner of (H). A dramatic reduction of p65 nuclear localisation is found with SB203580 treatment as detailed in the high magnification insert at the bottom right corner of (I). Indirect immunofluorescence; magnification ×600 (B–D), ×1200 (E–G), ×200 (H and I).
Effect of PA-LPS challenge on phosphorylation of tyrosine in airway epithelium
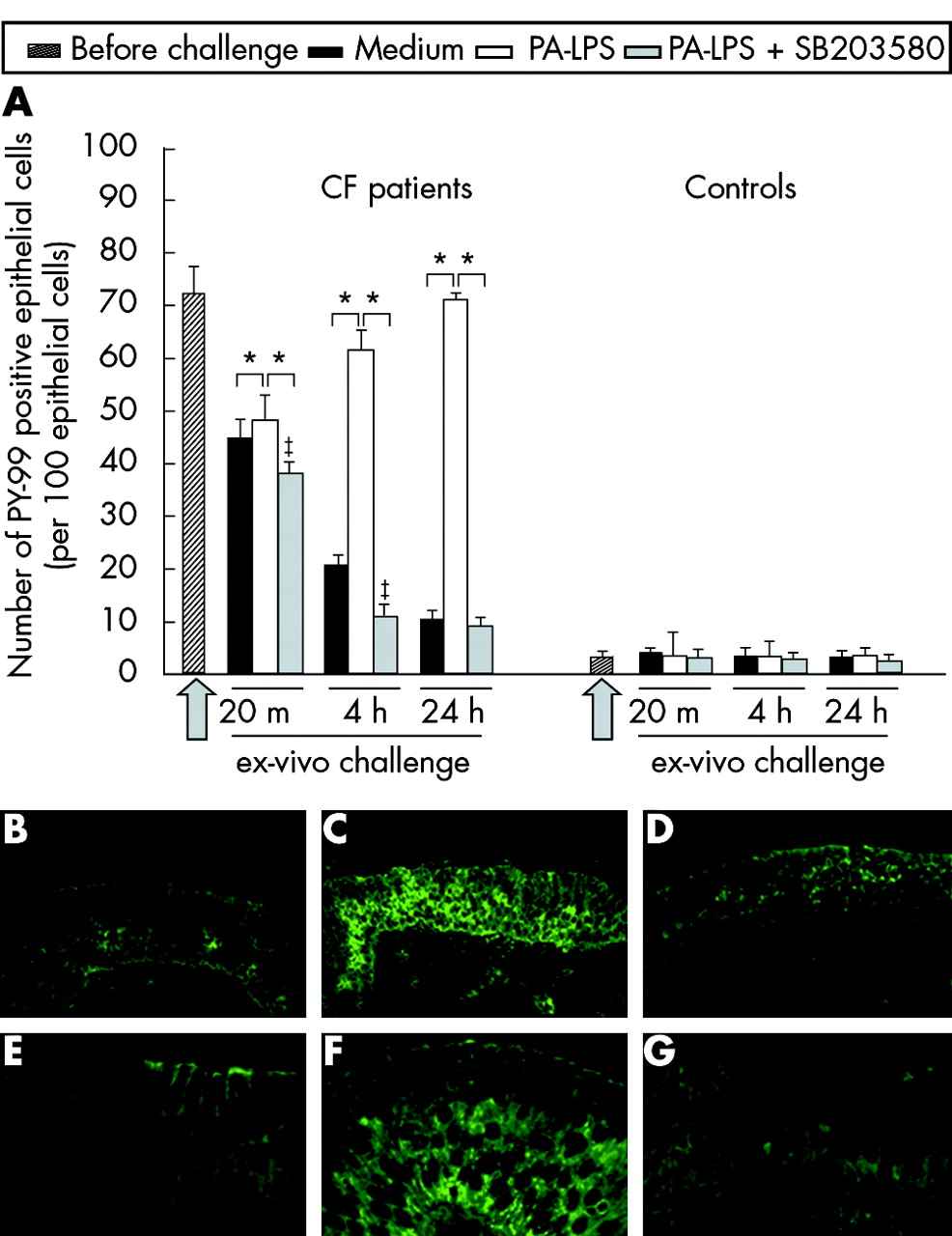
We also analysed phosphotyrosine expression by the epithelial cells in explanted nasal biopsy fragments stimulated with PA-LPS. After 4 hours of PA-LPS challenge a significant increase in tyrosine phosphorylation was seen in the epithelial compartment of CF polyps (p = 0.034; fig 2A, B, C, E and F) but not in controls (fig 2A). These changes were more evident after 24 hours of challenge and SB203580 treatment significantly inhibited epithelial phosphotyrosine induction (p = 0.017; fig 2A, D and G).

Upregulation of PY-99 expression by epithelial cells following 4 hours of PA-LPS challenge of nasal polyp explants from CF patients: effects of p38-MAP-k inhibition by SB203580. (A) Time course of PA-LPS induced PY-99 expression by nasal polyp epithelial cells in CF and control tissues and effect of p38-MAP-k inhibition by SB203580. Mean (SD) number of epithelial cells with PY-99 expression per 100 epithelial cells before challenge and after challenge with medium alone, PA-LPS, or PA-LPS + SB203580 following 20 minutes, 4 hours and 24 hours of incubation. The arrow indicates the start point of the challenge. *p <0.05 v samples cultured with medium alone and vs samples cultured with PA-LPS + SB203580; ‡p<0.05 v samples cultured with medium alone (Wilcoxon’s test). Combined data obtained from all seven CF or all seven control samples. (B)–(G) Phosphorylation of tyrosine in the airway epithelium, as revealed by PY-99 expression, in CF nasal polyp explants after 4 hours of challenge in the presence of medium alone (B and E), PA-LPS (C and F), and PA-LPS + SB203580 (D and G). Faint PY-99 epithelial expression is observed in the majority of epithelial cells after incubation with medium alone (B and E); PA-LPS challenge led to a dramatic increase in the number of epithelial cells with intense PY-99 expression (C and F) in the cytoplasm as well as on cell membranes (F), while treatment with SB203580 totally prevented PY-99 upregulation induced by PA-LPS challenge (D and G). Indirect immunofluorescence; magnification ×600 (E–G), ×200 (B–D).
Effect of PA-LPS challenge on mucosal inflammation
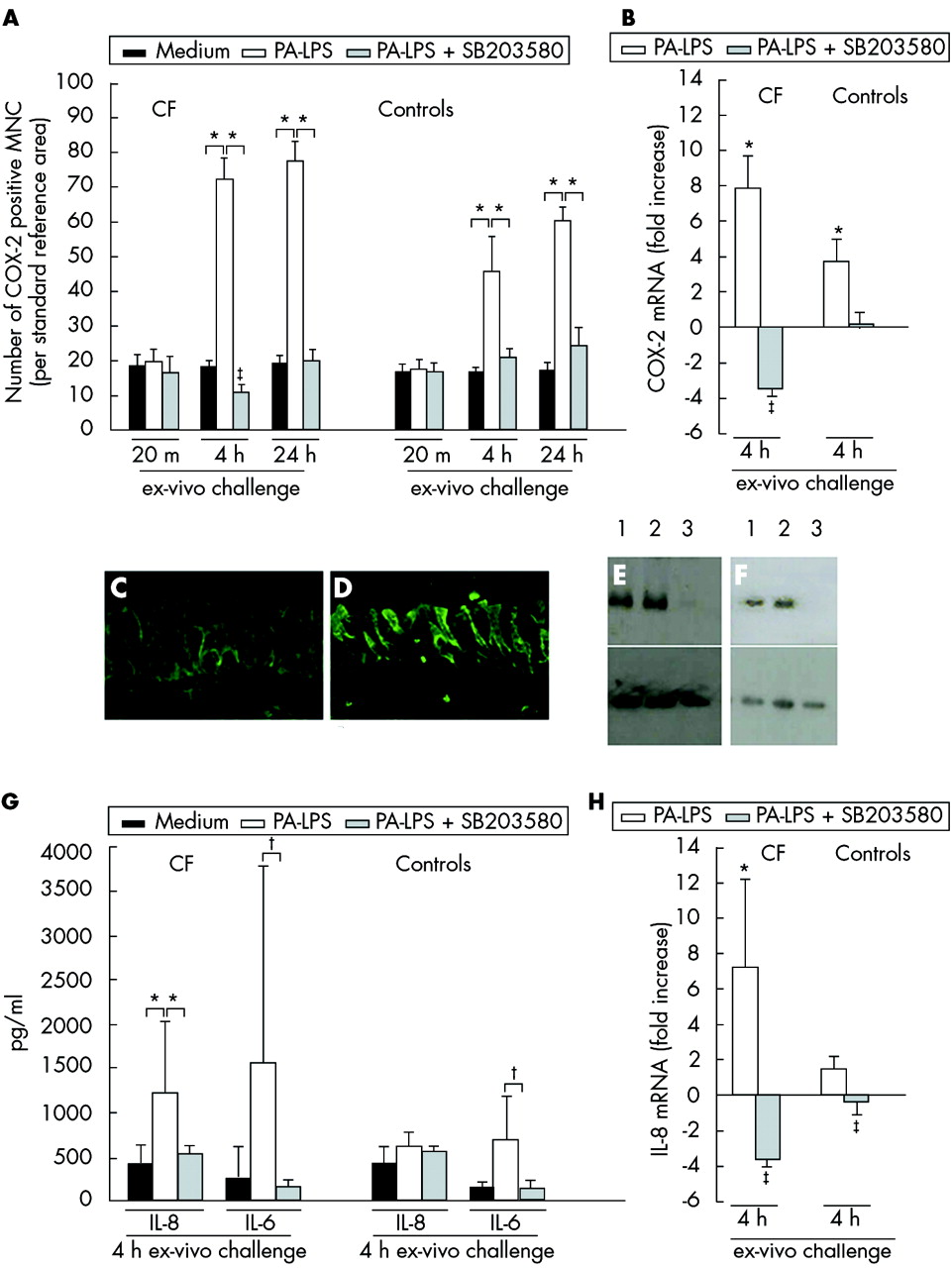
To study the inflammatory responses to PA-LPS challenge in CF nasal polyp explants, we also analysed the expression of COX-2 by mucosal mononuclear cells. This latter marker was studied as a broad parameter of inflammation as described in our previous study.13 After 4 hours of challenge and increasingly after 24 hours of incubation, PA-LPS also induced an increase in the numbers of mucosal COX-2 positive mononuclear cells (p = 0.043, fig 3A) and in the expression of COX-2 mRNA (p = 0.043, fig 3B) in both CF tissues and controls compared with samples cultured in medium alone. The increased expression of COX-2 following PA-LPS challenge was efficiently controlled by the p38-MAP-k inhibitor SB203580 (p = 0.043) which also reduced basal COX-2 expression observed in the absence of any LPS stimulation (p = 0.042, fig 3A and B).
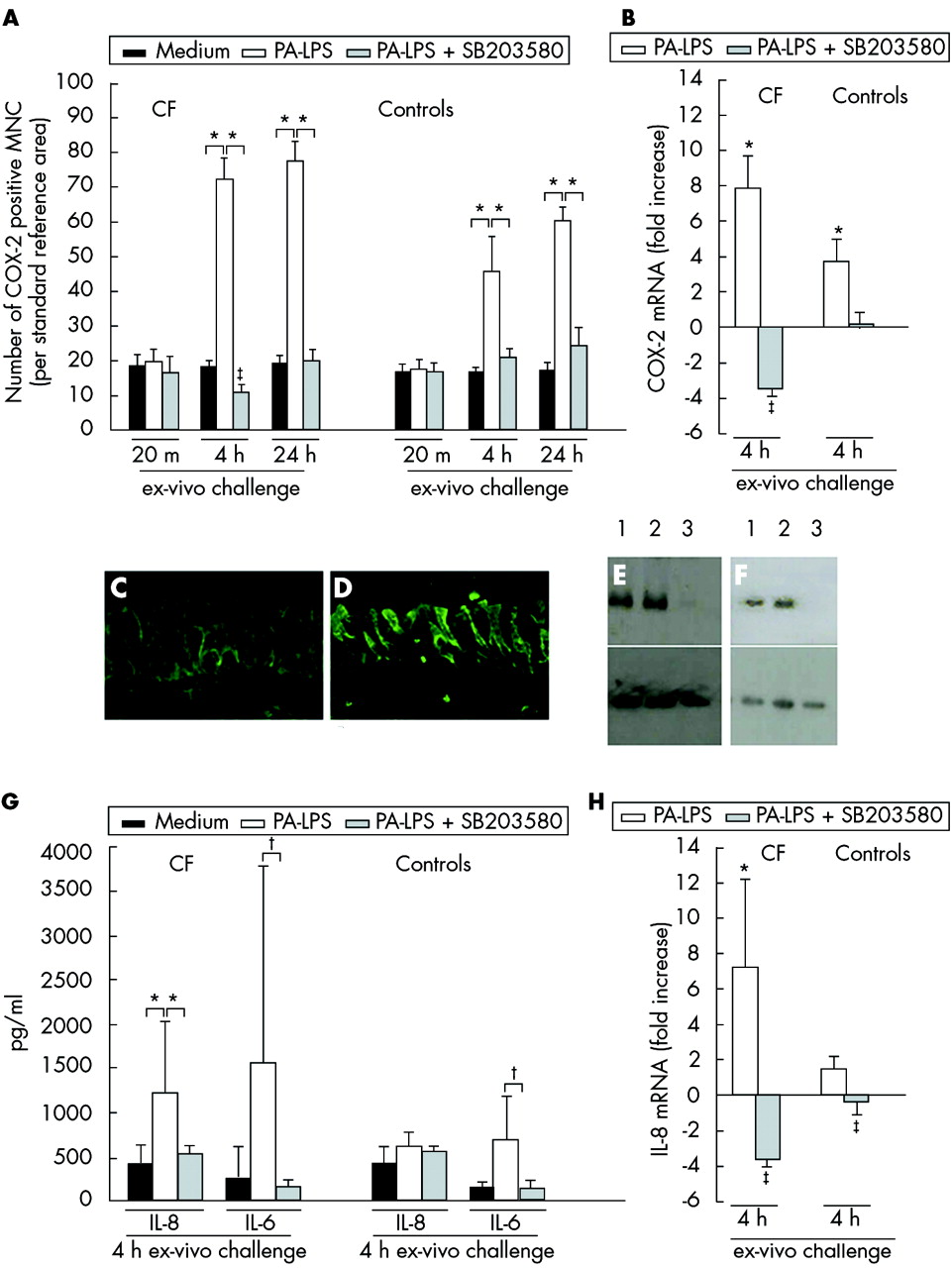
Effects of inhibition of the p38 MAP-k pathway in CF and control tissues. (A) Time course of COX-2 protein expression by mucosal mononuclear cells (MNC) following PA-LPS challenge of nasal polyp explants from CF patients and controls: effects of p-38-MAP-k inhibition by SB203580. Induction of COX-2 protein expression by mucosal MNC in all seven CF (left panel) and seven control (right panel) samples. Mean (SD) number of COX-2 positive MNC per mm2 of mucosa after challenge with medium alone, PA-LPS, or PA-LPS + SB203580 following 20 minutes, 4 hours, and 24 hours of incubation. *p<0.05 v samples cultured with medium alone and v samples cultured with PA-LPS + SB203580; ‡p<0.05 v samples cultured with medium alone (Wilcoxon’s test). (B) Effects on COX-2 mRNA levels after 4 hours of PA-LPS challenge of nasal polyp explants from CF patients and controls: effects of p-38-MAP-k inhibition by SB203580. Quantitative PCR of five CF and five control samples indicating induction of COX-2 mRNA by PA-LPS and the specific down modulation by the p38-MAP-k inhibitor SB203580 used at a final concentration of 1 μM/ml. Inhibition of the p38-MAP-k pathway reduced the level of COX-2 mRNA in CF tissues below the levels observed in the unstimulated but already inflamed biopsy tissue. *p<0.05 v samples cultured with medium alone and v samples cultured with PA-LPS + SB203580; ‡p<0.05 v samples cultured with medium alone (mean, SD; Wilcoxon’s test). (C), (D) p-38 MAP-k phosphorylation in epithelial cells after 4 hours of PA-LPS challenge of nasal polyp explants from CF patients in the presence of medium alone (C) and PA-LPS (D). Faint epithelial expression of phospho p38-MAP-k was observed after incubation with medium alone (C); the challenge with PA-LPS led to a dramatic increase in phospho p38-MAP-k expression in the cytoplasm as well as on cell membranes of many epithelial cells (D). Indirect immunofluorescence, magnification ×800 (C–D). (E), (F) Western blots of polyp explants of CF patient no 6 (E) and 7 (F) in table 2 after 4 hours of culture with medium alone (track 1), PA-LPS (track 2), and PA-LPS + SB203580 (track 3) as described in Methods section. Top panel shows phosphorylation of MAPKAP-k-2 at threonine 344 in presence of LPS as detected by the anti-MAPKAP-k-2 [pT334] polyclonal antibody. Almost complete inhibition was seen in samples cultured with PA-LPS + SB203580. Bottom panel shows that similar levels of actin were detected in all samples. (G) Effects on IL-8 and IL-6 protein expression in culture supernatants after 4 hours of PA-LPS challenge of nasal polyp explants from five CF patients and five controls: effects of p-38 MAP-k inhibition by SB203580. PA-LPS challenge induced an increase in IL-8 in all CF samples whereas IL-6 secretion was upregulated in four of five CF patients (lack of upregulation of IL-6 secretion in patient no 3). SB203580 significantly downregulated PA-LPS induced cytokine release. *p <0.05 v samples cultured with medium alone and v samples cultured with PA-LPS + SB203580; †p = 0.08; ‡p<0.05 v samples cultured with medium alone (mean, SD; Wilcoxon’s test). (H) Effects on IL-8 mRNA levels in cultured mucosal explants after 4 hours of PA-LPS challenge of nasal polyp explants from CF patients and controls: effects of p-38-MAP-k inhibition by SB203580. Quantitative PCR of five CF patients and five controls indicating induction of IL-8 mRNA by PA-LPS and specific down modulation by SB203580 (1 μM/ml). Inhibition of the p38-MAP-k pathway reduced the level of IL-8 mRNA in CF tissues below the levels observed in the unstimulated but already inflamed biopsy. *p<0.05 v samples cultured with medium alone and v samples cultured with PA-LPS + SB203580; ‡p<0.05 v samples cultured with medium alone (mean, SD; Wilcoxon’s test).
Effect of PA-LPS challenge on activation of p38 in the airway epithelium and of SB203580 on p38-MAP-k activity
The results so far described suggest involvement of p38-MAP-k. We therefore studied p38-MAP-k phosphorylation in the epithelial cells after PA-LPS challenge. The mean (SD) number of phospho-p38 positive epithelial cells per 100 epithelial cells after 4 hours of challenge was 49.4 (2.8) in CF tissues v 11.2 (1.0) after culture with medium (p<0.03) and 4.3 (1.9) v 3.7 (1.6) in control tissues (p>0.5; fig 3C, D). To test whether SB203580 inhibited p38-MAP-k activity we monitored the phosphorylation of MAPKAP-k-2, a specific p38-MAP-k substrate.23 The data shown in fig 3E and 3F indicate that SB203580 controlled phosphorylation of MAPKAP-k-2 at threonine 334. These results suggest that p38-MAP-k is activated in CF tissues and, in our model, SB203580 specifically inhibited the downstream effects of this activation.
Effect of PA-LPS challenge on IL-8 upregulation and neutrophil recruitment to the upper mucosal compartments in CF tissues
One of the key aims of this study was to establish whether IL-8 production and leucocyte migration in CF are controlled by p38-MAP-k inhibition, since neutrophil containment is a target of CF treatment. IL-8 levels were already significantly increased in culture supernatants from all CF samples after 4 hours of challenge with PA-LPS compared with medium alone (p = 0.043, fig 3G). IL-6 was also generally increased but the difference did not reach statistical significance because in one patient (patient no 3, table 1) there was no increase in IL-6 release following PA-LPS challenge (fig 3G). No differences in IL-1β, IL-10, TNF-α, or IL-12p70 expression were found (data not shown).
IL-8 mRNA was significantly increased in CF tissues after 4 hours of PA-LPS challenge compared with samples cultured with medium alone (p = 0.043, fig 3H). The addition of SB203580 completely inhibited the increase in IL-8 mRNA (p = 0.043) as well as IL-8 release (p = 0.043, fig 3G and H) and also partially controlled IL-6 upregulation (p = 0.08, fig 3G). Controls only showed minimal IL-8 upregulation and release of IL-8 or IL-6 (fig 3G and H).
After 24 hours PA-LPS induced a remarkable modification in the pattern of neutrophil distribution in the biopsies in all CF samples, but not in controls. Neutrophils migrated from the deeper mucosal layers and accumulated in the upper (subepithelial and intraepithelial) mucosal compartments (fig 4A, B, C, E) with clustering, degranulation, and release of cathepsin G in the extracellular matrix determined as described in the legend to fig 4 and in the online supplement (p = 0.018 v samples challenged with medium alone for neutrophil redistribution). Neutrophils also crossed the epithelium and moved to the luminal part of the biopsies where clusters of neutrophils were evident outside the epithelial apical membrane (fig 4A, C, E). No changes in neutrophil distribution in the tissue were observed after 20 minutes or 4 hours (data not shown). SB203580 treatment abolished these PA-LPS induced changes (p = 0.018 v samples challenged with PA-LPS alone). In the presence of SB203580 the majority of mucosal neutrophils seeded within the deeper mucosal layers, and the numbers of neutrophils within the subepithelial and intraepithelial mucosal compartments as well as outside the epithelial apical membrane were similar to those observed after culture with medium alone (fig 4A, D, F).

Neutrophil recruitment from the deeper mucosal layers to the mucosal surface after 24 hours of PA-LPS challenge of nasal polyp explants from CF and control patients: effects of p38-MAP-k inhibition by SB203580. (A) Mean (SD) number of neutrophils (cathepsin G positive PMN leucocytes) in different mucosal compartments in all seven CF and control samples; dl, deeper mucosal layers (per reference area of 5×104 μm2 from bottom of the mucosa to 100 μm below the epithelial basal membrane); sec, subepithelial region (per reference area of 5×104 μm2, 100 μm thick below the epithelial basal membrane); iec, within the epithelium (per mm of epithelium); lumen, in the area outside the epithelial apical membrane (per reference area of 2.5×104 μm2, 50 μm thick just above the epithelial surface). *p <0.05 (Wilcoxon’s test). (B)–(F) In the absence of any antigen stimulation, no accumulation of neutrophils (labelled by anti-cathepsin G monoclonal antibody) was evident in the subepithelial compartment and only a few neutrophils were detected within the epithelium. No degranulation of neutrophils or presence of neutrophils at the luminal side was evident (B). PA-LPS challenge induced a dramatic modification in leucocyte distribution: neutrophils migrated from deeper mucosal layers and accumulated in the upper (subepithelial and intraepithelial) mucosal compartments with clustering, degranulation, and release of cathepsin G in the extracellular matrix. Neutrophils crossed the epithelial basal membrane, induced derangement of the epithelial morphology, and moved to the luminal part of the biopsy where clusters of neutrophils were evident outside the epithelial apical membrane (C and E, high magnification of C). The inset in the top right corner of E details a cluster of neutrophils outside the epithelial apical membrane as indicated by the arrow. Treatment with SB203580 + PA-LPS totally prevented degranulation and migration of neutrophils induced by PA-LPS: no accumulation of neutrophils was evident at the subepithelial level, and no epithelial infiltration with derangement of mucosal morphology or migration outside the epithelial apical membrane was observed (D and F, high magnification of D). The latter pattern is similar to that observed in the absence of PA-LPS stimulation. Indirect immunoperoxidase staining with anti-cathepsin G monoclonal antibody, haematoxylin counterstain. Magnification ×200 (B–D), ×600 (E–F).
We also tested in vitro neutrophil migration in the presence or absence of the SB203580 inhibitor to define whether the observed neutrophil inhibition operated by SB203580 in the cultured nasal polyp mucosa was due to IL-8 inhibition or to a direct effect on neutrophils. The data shown in table 2 and in fig 5 indicate that SB203580 did not hinder migration but paradoxically favoured neutrophil migration.
Results of in vitro neutrophil migration in the presence of SB 203580

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Example of results obtained using the MIGRATEST assay. (A) Migration of neutrophils in the absence of a stimulus, (B) migration of neutrophils in the presence of fMLP, and (C) migration of neutrophils pretreated with 1 μM SB203580 in the presence of fMLP. Top quadrants: red dots represent neutrophils; bottom quadrants: blue dots represent calibrating beads. In control samples (A and B) 1/1000 DMSO was added to exclude any non-specific effect in the SB203580 (diluted in DMSO) treated sample.
These results suggest that PA-LPS challenge induces IL-8 upregulation and neutrophil migration to the upper mucosa layers in CF tissue but not in control tissue. They also suggest that IL-8 inhibition is the primary cause of the changes in neutrophil distribution.
DISCUSSION
Control of inflammation is a key objective in the treatment of CF, as migration of neutrophils into the open air spaces and their degranulation contribute to irreversible lung damage and death of patients with CF.1 Of all the inflammatory cytokines/chemokines, IL-8 is considered to be the main player in orchestrating inflammation in CF.1 Importantly, IL-8 has a direct effect on neutrophils by controlling and directing their migratory capacities as well as their activation.24 Other inflammatory cytokines are also increased in CF but their contribution to CF inflammation is not fully understood. It is commonly accepted that infections take advantage of the “fertile” CF hyperresponsive state to sustain chronic inflammation and cause tissue damage in patients with CF.1,25 The importance of environmental factors in sustaining the inflammatory response is supported by our data as a decrease in p65 nuclear localisation and phosphotyrosine expression is observed when explants are cultured in medium alone. However, we observed that CF tissue was more reactive to PA-LPS, as shown in fig 4 where leucocyte migration to the upper mucosal layers was only seen in CF tissue.
It has recently been reported that activation of the p38-MAP-k pathway is required for Burkholderia cepacia (BC) induced IL-8 release.16 We adopted a similar methodology to that study using PA-LPS as an environmental stimulus to induce inflammation and trigger IL-8 production.16 While release of IL-8 was highly controlled by p38-MAP-k inhibition, other signal transduction pathways such as NF-κB has been found to be involved in this process.5 However, in our study we detected a reduction in NF-κB activation after p38-MAP-k inhibition (fig 1), indicating a potential extra beneficial effect of neutralisation of this signal transduction pathway. Recent reports have indicated that SB203580 affects NF-κB signalling in intricate ways. It has been shown that SB203580 inhibits p65 phosphorylation,26,27 but in our model the most obvious effect of SB203580 was the inhibition of p65 nuclear translocation.28 Inhibition of the p38-MAP-k pathway might also directly affect neutrophil function.18,29 Our data, which indicate inhibition of neutrophil migration by SB203580, might be the result of a combination of indirect (via inhibition of IL-8 secretion, for example) and direct effects on neutrophils. Since neutrophils might release IL-8, SB203580 would thus interfere with a possible self-feeding pathogenic loop. Our experiments demonstrate a specific reduction in IL-8 secretion, supporting the view that epithelial activation with IL-8 secretion precedes neutrophil migration and activation. As indicated above, other cells present in the mucosa might also be involved. In this context it has recently been reported that monocytes from CF patients, heterozygous or homozygous for CFTR mutations, produce more IL-8 when stimulated with LPS.30 In that report it was shown that the p38-MAP-k pathway was also involved and that the increased responsiveness was not related to upregulation of LPS receptor.30 These results help to explain our observation of a good response to LPS challenge in CF tissue but not in controls. Given that PA and BC mediated IL-8 induction is under the control of the p38-MAP-k pathways, and that even “sterile” hyperosmotic stimulation induces IL-8 release controlled by the activation of p38-MAP-k,15 the inhibition of this signal transduction pathway may represent a valid therapeutic target to control IL-8 production, neutrophil recruitment,31 and activation18 and, ultimately, inflammation in CF. The data presented here demonstrate the efficacy of p38-MAP-k inhibition with a significant decrease in IL-8 production (fig 3). The most compelling result is the effect on neutrophil migration and degranulation as shown in fig 4.
The significance of tyrosine phosphorylation following LPS stimulation and the inhibition of this response by the p38-MAP-k inhibitor is, however, unclear. We have not identified which proteins are tyrosine phosphorylated or the potential downstream tyrosine kinase involved. However, inhibition of tyrosine phosphorylation by the p38-MAP-k inhibitor is consistent with the more specific reductions in COX-2 and IL-8 expression we observed. Further work will be required to clarify which tyrosine kinases are influenced.
Previous studies in animal models of acute respiratory inflammation have shown that inhibition of p38-MAP-k reduces neutrophil involvement in respiratory inflammation.18,32,33 Our report validates this approach in humans using CF tissue and exploits the ex vivo organ culture model. This model complements and expands studies on isolated single cell types (such as epithelial cells). The validity of ex vivo organ cultures has been highlighted by studies in other chronic inflammatory diseases where they have helped to clarify critical pathogenic steps and outline possible new therapeutic strategies.13 Our study has been performed in a small group of patients but the results we obtained have been clearcut, consistent in all samples, and highly statistically significant.
The inhibition of neutrophil migration and degranulation seen in our studies is significant for patients with CF since neutrophil containment is a primary target in CF treatment. In spite of the limited viability of p38-MAP-k deficient mice,34 the clinical use of p38-MAP-k inhibitors is considered to be an attractive therapeutic target to control chronic inflammatory diseases35 and even cancer.36 Thus, new generations of p38-MAP-k inhibitors are under development and are currently being used in ongoing clinical trials.37,38 A major opportunity in CF could lie in the topical application of such inhibitors via aerosol to target lung inflammation, thus reducing the potential systemic side effects.
We have provided a rationale and proof of principle for the use of p38-MAP-k inhibitors to control CF inflammation, but we have to consider the limitations of the use of nasal polyps to support our conclusions. Although tissue from nasal polyps is useful to study ex vivo CF inflamamtion and its modulation, only a selected and not a fully representative group of patients is studied. Despite this limitation, we believe that studies using nasal polyps are valuable to test potential new therapeutic strategies in CF. At present many compounds with these characteristics are undergoing clinical trials for other (non-CF) inflammatory conditions of the upper respiratory tract.39 The use of p38-MAP-k inhibitors may be an attractive option in the treatment of CF.
REFERENCES
Supplementary materials
The report is available as a downloadable PDF (printer friendly file).
If you do not have Adobe Reader installed on your computer,
you can download this free-of-charge, please Click hereFiles in this Data Supplement:
- [view PDF] - Inhibition of p38 Mitogen-Activated-Protein-Kinase controls airway inflammation in cystic fibrosis - online only and supplemental report.
Footnotes
-
Published Online First 30 June 2005
-
Funded by ARC-UK and ICH grants and the Italian Cystic Fibrosis Research Foundation, FFC Latina, grant #16/2004, CICRA UK, Cancer Research UK.
-
Competing interests: none declared.