Article Text

Abstract
Background: The role of transforming growth factor-β1 (TGF-β1) in chronic obstructive pulmonary disease is still controversial, but it has been proposed that it may protect from mucus hypersecretion since it is able to downregulate mucin production. A study was undertaken to investigate the expression of TGF-β1 and its type II receptor (TGF-β RII) in the bronchial glands of smokers with COPD.
Methods: The expression of TGF-β1 and TGF-β RII were examined immunohistochemically in the bronchial glands of 24 smokers undergoing lung resection for solitary peripheral nodules: 12 with airflow limitation (smokers with COPD) and 12 with normal lung function.
Results: The expression of TGF-β1 in bronchial glands was similar in the two groups of subjects while that of TGF-β RII was lower in smokers with COPD than in smokers with normal lung function (p = 0.004). TGF-β RII expression was inversely correlated with the values of Reid’s index, a measure of gland size (p = 0.02, r = −0.50).
Conclusions: In the bronchial glands of smokers with COPD there is decreased expression of TGF-β RII which is associated with bronchial gland enlargement. These findings support the view that the absence of TGF-β signalling may induce structural changes in the bronchial glands which, in turn, may promote mucus hypersecretion.
- FEV1, forced expiratory volume in 1 second
- FVC, forced vital capacity
- Pao2, Paco2, arterial oxygen and carbon dioxide tensions
- TGF-β, transforming growth factor β
- TGF-β RII, TGF-β type II receptor
- chronic obstructive pulmonary disease
- transforming growth factor β (TGF-β)
- transforming growth factor type II receptor (TGF-β RII)
- mucus hypersecretion
Statistics from Altmetric.com
- FEV1, forced expiratory volume in 1 second
- FVC, forced vital capacity
- Pao2, Paco2, arterial oxygen and carbon dioxide tensions
- TGF-β, transforming growth factor β
- TGF-β RII, TGF-β type II receptor
- chronic obstructive pulmonary disease
- transforming growth factor β (TGF-β)
- transforming growth factor type II receptor (TGF-β RII)
- mucus hypersecretion
Chronic obstructive pulmonary disease (COPD) is a public health problem worldwide, being a major cause of chronic morbidity and mortality.1,2 Although it is well established that cigarette smoking is the main risk factor for the development of COPD, the pathogenesis of the disease is still incompletely understood.3,4 It has recently been suggested that transforming growth factor β (TGF-β) may be involved in the development of COPD,1 even if its role remains controversial.
In the past there has been considerable interest in TGF-β1, with a few studies reporting an increased expression of this growth factor in the airways of smokers with COPD.5–7 It was therefore proposed that TGF-β, through fibrosis and thickening of the airway wall, may contribute to the development of airflow limitation in smokers. However, more recently, several studies have pointed out that TGF-β1 may have a protective role as well. Indeed, in animal models it has been clearly shown that alterations that abrogate TGF-β1 signalling result in an increased risk of pulmonary emphysema.8,9 In humans, genetic studies have shown that polymorphisms of the TGF-β1 gene associated with a higher production of this growth factor are less common in smokers with airflow limitation than in healthy smokers, suggesting that TGF-β1 may protect against the development of COPD.10,11
One of the possible mechanisms by which TGF-β1 may exert a protective role is by switching off the inflammatory response, preventing its perpetuation. Indeed, it has been shown that TGF-β1 orchestrates the resolution of inflammatory responses, acting on both T and B lymphocytes.12–14 Moreover, TGF-β1 attenuates the response to viruses and bacteria by inhibiting the upregulation of mucins induced by activation of the Toll like receptors.15 In this context, it has been reported that TGF-β1 is crucial for downregulating mucin production after infection with Haemophilus influenzae, which is a common pathogen in COPD.16
While the role of TGF-β1 signalling on mucin production has been investigated in vitro, to our knowledge no study has addressed the relevance of the TGF-β1 pathway on mucus hypersecretion in vivo. In a previous study we have shown that, in smokers with COPD, mucus hypersecretion was associated with an inflammatory process localised within the bronchial glands.17 In this study we aimed to extend those findings by analysing the expression of a cytokine involved in mucus production such as TGF-β1, specifically in the gland compartment.
The expression of TGF-β1 and TGF-β type II receptor (TGF-β RII) in the bronchial glands of 24 smokers undergoing lung resection for solitary peripheral nodules was examined. Twelve patients had COPD and 12 had normal lung function. Even though we had tissue available from only three non-smoking subjects, we decided also to quantify the expression of TGF-β1 and its receptor in these subjects for comparison purposes. Some of the results of this study have been presented in abstract form elsewhere.18,19
METHODS
Subjects
The study population comprised 24 subjects who underwent lung resection for a solitary peripheral carcinoma. All had a history of cigarette smoking; 12 had airflow limitation with a ratio of forced expiratory in 1 second to forced vital capacity (FEV1/FVC) <70% after bronchodilator inhalation (smokers with COPD) and 12 were asymptomatic with normal lung function (control smokers). Ten of the patients with COPD had symptoms of chronic bronchitis, defined as cough and sputum production occurring on most days of the month for at least 3 months a year during the 2 years prior to the study. Subjects with COPD had no exacerbations (defined as increased dyspnoea associated with a change in the quality and quantity of sputum) that led the subject to seek medical attention during the month preceding the study.
For comparison purposes we also included in the analysis specimens obtained from three non-smoking subjects who underwent lung resection for a solitary peripheral carcinoma. They had no symptoms of chronic bronchitis or airflow limitation (mean (SD) FEV1/FVC 81 (8)%).
All subjects included in the study had been free of acute upper respiratory tract infections and none had received corticosteroids or antibiotics during the month preceding surgery or bronchodilators within the previous 48 hours. The subjects were non-atopic (negative skin tests to common allergen extracts) and had no past history of asthma or allergic rhinitis. Each subject underwent an interview, chest radiography, electrocardiography, routine blood tests, skin tests with common allergen extracts, and pulmonary function tests in the week before surgery. Some of the subjects in this study were included in a previous report.17
The study conformed to the Declaration of Helsinki and informed written consent was obtained for each subject undergoing surgery.
Pulmonary function tests
Pulmonary function tests including measurements of FEV1 and FVC were performed as previously described17 in all the subjects examined. The predicted normal values used were those from Communité Europeenne du Carbon e de l’Acier (CECA).20 In order to assess the reversibility of airway obstruction in subjects with COPD, the pulmonary function measurement was repeated 15 minutes after inhalation of 200 μg salbutamol. Subjects with normal lung function underwent inhalation challenge with methacholine.
Histology
Bronchial rings were taken from the lobar or segmental bronchus of the lobe obtained at surgery, away from the tumour site. One bronchial ring was selected for each subject, fixed in 4% formaldehyde and, after dehydration, embedded in paraffin wax. Bronchial rings were then orientated and 5 μm thick serial sections were cut for immunohistochemical analysis of TGF-β1 and TGF-β RII expression. Briefly, sections were subjected to antigen retrieval to unmask antigens in formalin fixed and paraffin embedded tissue sections by heating in a microwave oven on high power for 8 minutes in 0.01 mol/l citrate buffer (pH 6.0) and then incubated with a mouse monoclonal antibody anti-TGF-β1 (dilution 1:20; Genzyme Diagnostics, Cambridge, MA, USA) or with a polyclonal antibody anti-TGF-β RII (dilution 1:200; Biotechnology Inc, Santa Cruz, CA, USA). Before incubation with primary antibody the sections were treated with a biotin blocking kit (Vector Laboratories, Peterborough, UK) to inhibit endogenous biotin. The detection system was performed using the Vectastain ABC kit (Vector Laboratories) with 3-amino-9-ethylcarbazole as the chromogenic substrate. Sections were counterstained with Mayer’s haematoxylin.
Morphometric measurements were performed by a single observer using a computerised image analyser (Casti Imaging SC processing, Venice, Italy). Briefly, in each patient the total gland area was identified at lower magnification (20×) and outlined with the computer cursor. Then, at higher magnification (40×), the number of acini positive for TGF-β1 or TGF-β RII staining was quantified by the observer and the results were expressed as a percentage of the total number of acini. For an acinus to be positive, specific staining had to be present on at least a quarter of the total acinus. In addition, the area positive for TGF-β1 or TGF-β RII staining was outlined with the computer cursor in each acinus and the sum of these areas was computed by the image system. The results were expressed as percentage of positive area over the total gland area.
Reid’s index, which measures bronchial gland size, was calculated by computing the ratio between the maximum thickness of each bronchial gland and the thickness of the bronchial wall.17 Finally, to quantify mucin producing acini, Alcian blue-Periodic Acid Schiff (PAS) staining was performed which identifies acidic and neutral mucopolysaccharides, respectively. The results were expressed as a ratio between Alcian blue-PAS− and Alcian blue-PAS+ acini, which thereafter will be referred to as mucin negative and mucin positive acini.
Statistical analysis
Group data were expressed as mean (SE) or as medians with ranges when appropriate. Differences between groups were analysed with the unpaired Student’s t test for clinical data and the Mann-Whitney U test for morphological data. Correlation coefficients were calculated using Spearman’s rank method. Values of p<0.05 were considered significant. At least three replicate measurements of morphometric parameters were performed by the same observer and the intraobserver variability was assessed with the coefficient of variation (CV) for repeated measurements. For each subject CV was calculated as the ratio between the standard deviation (SD) and the mean of the three measurements performed: (SD/mean) × 100.
RESULTS
Clinical findings
The characteristics of the smokers with COPD and control smokers are shown in table 1. The two groups of subjects were similar with regard to age, sex, and smoking history (pack years). There were seven ex-smokers among the smokers with COPD and eight in the control smoker group. As expected from the selection criteria, smokers with COPD had a significantly lower value of FEV1 (% predicted) and FEV1/FVC ratio (%) than did control smokers, while the two groups of subjects were similar with regard to arterial oxygen and carbon dioxide tensions (Pao2 and Paco2; table 1). According to the GOLD criteria, three smokers with COPD were classified as stage 1 and nine as stage 2.1 All control smokers had normal lung function and reactivity to methacholine was within the normal range (PD20 FEV1 >1.44 mg methacholine, corresponding to 7 μmol).
Characteristics of smokers with COPD and control smokers
Histological findings
Quantification of TGF-β1 and TGF-β RII was satisfactory in all subjects included in the study except in one control smoker in whom quantification of TGF-β RII could not be performed because only a small amount of tissue was available. The total number of acini examined did not differ between smokers with COPD and control smokers (median (range) 172 (52–386) v 122 (55–384)). Similarly, the total gland area did not differ between the two groups (1 (0.4–3.0) mm2v 0.6 (0.3–2.0) mm2).
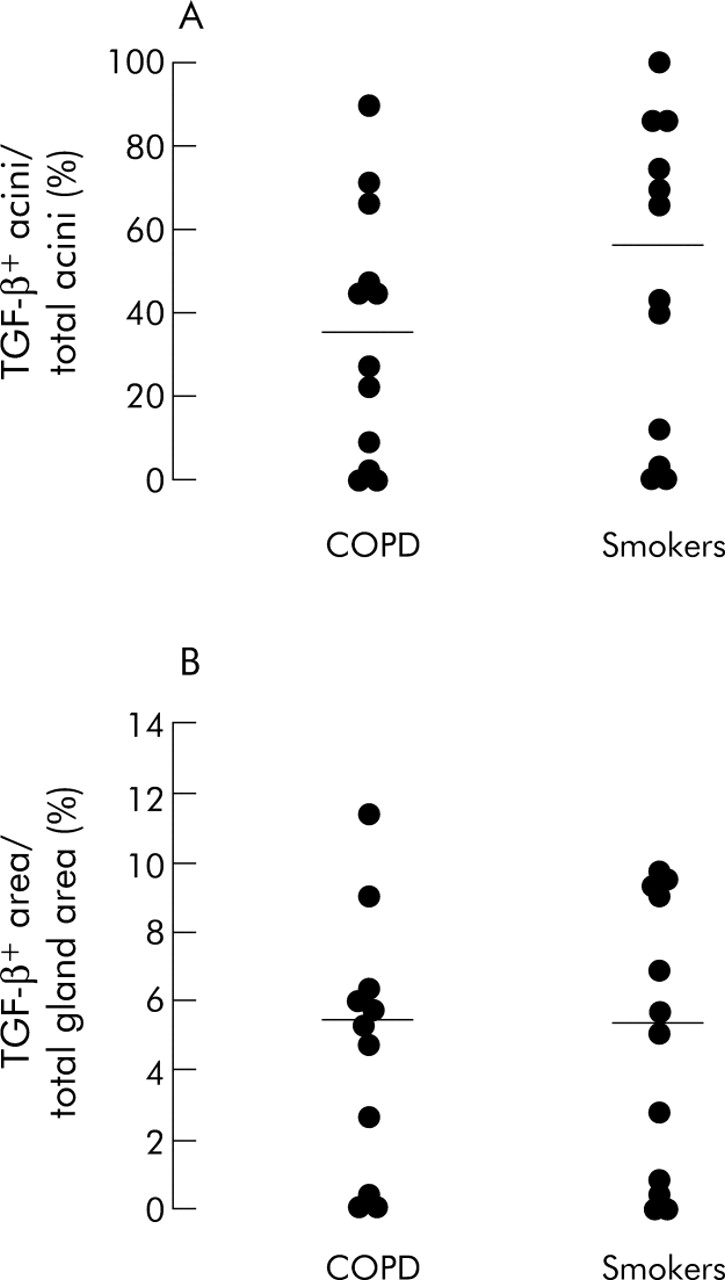
The percentage of TGF-β RII+ acini over total acini was lower in smokers with COPD than in control smokers (48 (7–90)% v 69 (54–100)%; p = 0.004)). When the results were expressed as percentage of positive area over total gland area, smokers with COPD had a lower percentage of TGF-β RII+ area than control smokers (7 (2–12)% v 11 (5–22)%; p = 0.012, figs 1 and 2). By contrast, the expression of TGF-β1 was not significantly different between smokers with COPD and control smokers, either when the results were expressed as percentage of positive acini or as percentage of positive area (fig 3).

Individual counts of (A) TGF-β RII+ acini (% total acini) and (B) TGF-β RII+ area (% total gland area) in the bronchial glands of smokers with COPD and control smokers. Horizontal bars represent median values.
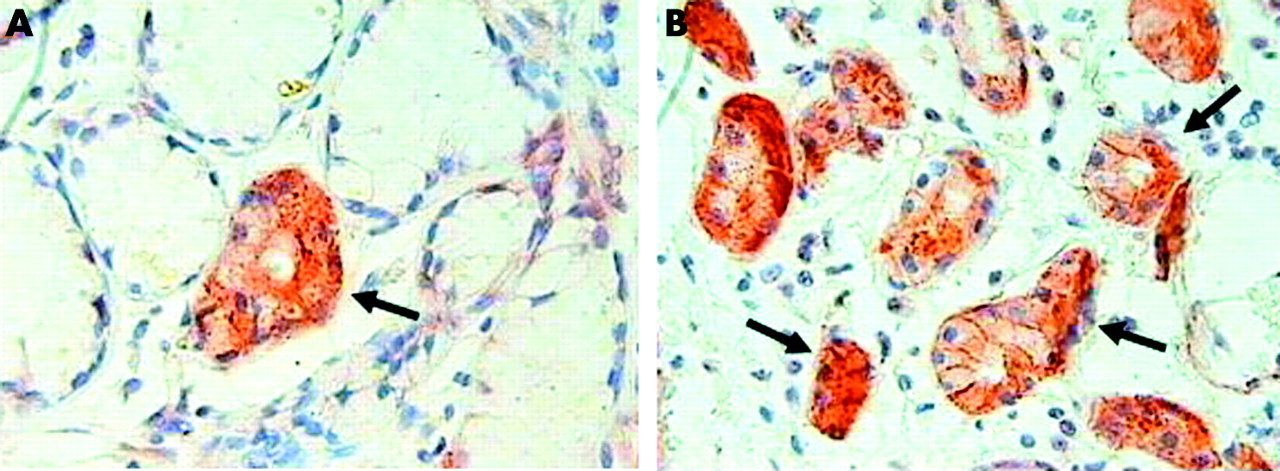
Micrograph showing TGF-β RII+ acini in bronchial glands of (A) a smoker with COPD and (B) a control smoker. Arrows indicate TGF-β RII+ acini which are stained brown. Original magnification ×630.

Individual counts of (A) TGF-β1+ acini (% total acini) and (B) TGF-β1+ area (% total gland area) in the bronchial glands of COPD and control smokers. Horizontal bars represent median values.
When the subjects were subdivided according to the presence or absence of symptoms of chronic bronchitis (instead of airflow limitation), TGF-β RII expression in smokers with chronic bronchitis (n = 10) was decreased compared with smokers without chronic bronchitis (n = 14). Indeed, patients with chronic bronchitis had a lower percentage of TGF-β RII+ acini than smokers without chronic bronchitis (48 (32–90)% v 66 (7–100)%; p = 0.01) as well as a lower percentage of TGF-β RII+ area (7 (3–10)% v 11 (2–22)%; p = 0.01). When the subjects were subdivided according to their current smoking status, no significant difference was observed between current smokers (n = 9) and ex-smokers (n = 15) in the percentage of TGF-β RII+ area (6 (2–22)% v 9 (3–18)%) or TGF-β1+ area (5 (0–11)% v 6 (0–16)%). In the three non-smoking subjects the values of TGF-β RII and TGF-β1 expression were in the same range as control smokers with a median (range) TGF-β RII+ area of 12 (10–12)% and TGF-β1+ area of 13 (6–16)%.
Bronchial gland size, as expressed by Reid’s index, was not significantly different between smokers with COPD and control smokers (45 (27–64)% v 38 (17–76%)). Similarly, the ratio between mucin negative and mucin positive acini was not significantly different between the two groups of subjects (9 (0–55)% v 6 (0–97)%). However, when all the smoking subjects were considered together, both the percentage of TGF-β RII+ acini and the percentage of TGF-β RII+ area were negatively correlated with the values of Reid’s index (p = 0.02, r = −0.50 and p = 0.01, r = −0.54, respectively; fig 4). Similarly, a trend towards a significant correlation was observed between the percentage of TGF-β RII+ acini and the ratio between mucin negative and mucin positive acini (p = 0.06, r = 0.40). Mucin positive acini were identified in sections stained with Alcian blue-PAS staining where they can be stained either in blue (acidic mucopolysaccharides) or purple (neutral mucopolysaccharides; fig 5). In serial sections, expression of TGF-β1 and TGF-β RII was not restricted to a particular type of acinus but was observed in both mucin positive and mucin negative acini.
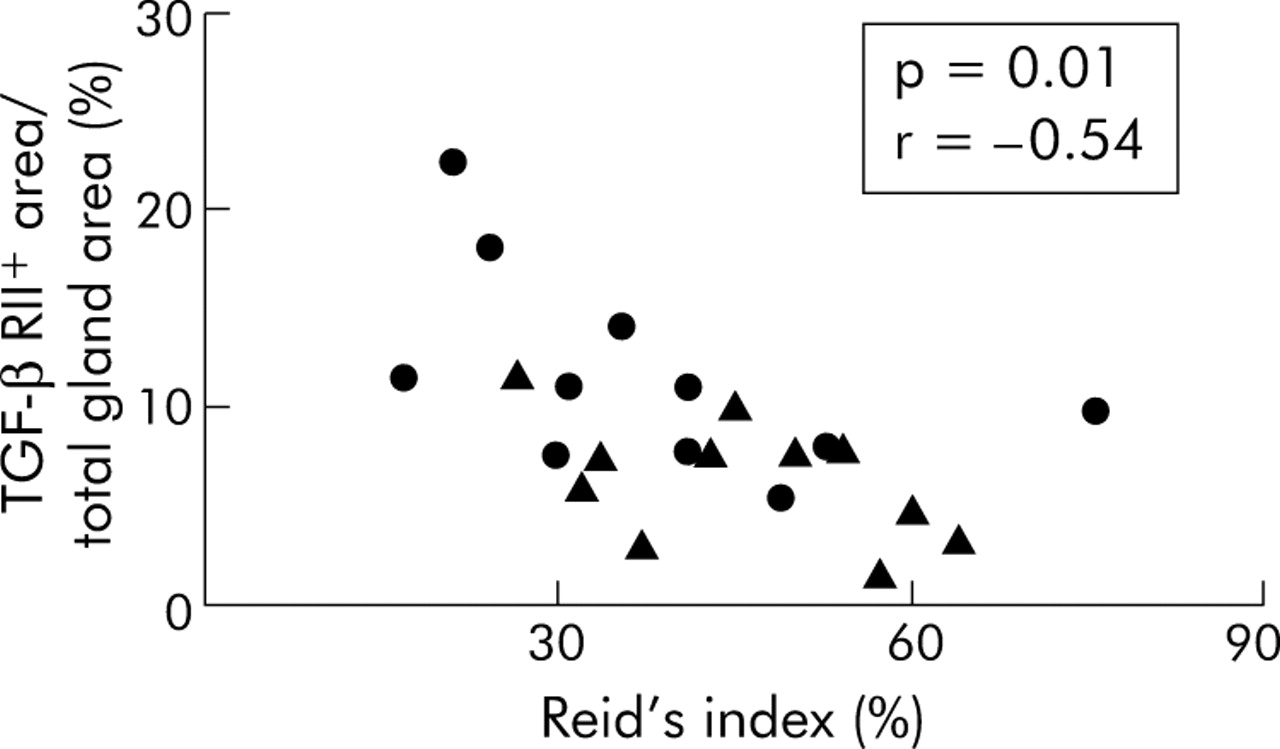
Relationship between TGF-β RII+ area (% total gland area) and Reid’s index (%); Spearman’s rank correlation, p = 0.01, r = −0.54. Smokers with COPD indicated by triangles and control smokers by circles.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Micrograph showing Alcian blue-PAS staining in a bronchial gland of a smoker with COPD. Mucin positive acini can be stained either in blue (acidic mucopolysaccharides) or purple (neutral mucopolysaccharides) as indicated by the arrows, while mucin negative acini are indicated by the arrowhead. Original magnification ×400.
The number of neutrophils infiltrating the bronchial glands was increased in smokers with COPD compared with control smokers (42 (12–88) v 11 (5–57) cells/mm2; p = 0.01), but this neutrophilic infiltration was not significantly correlated with either TGF-β1 or TGF-β RII expression.
When TGF-β1 and TGF-β RII expression in the bronchial epithelium was examined, these proteins were present at extremely high levels in both ciliated and goblet cells in smokers with COPD, control smokers, and even in non-smokers. For this reason a detailed quantification in the epithelium could not be performed.
The mean CV for repeated measurements were 2.9% for TGF-β RII+ acini; 1.3% for TGF-β RII+ area; 2.7% TGF-β1+ acini; and 11% for TGF-β1+ area.
DISCUSSION
This study shows that, in smokers with COPD, there is a decreased expression of TGF-β RII in the bronchial glands which is associated with bronchial gland enlargement. This suggests that impaired TGF-β signalling might induce structural changes in the bronchial glands which, in turn, may promote mucus hypersecretion.
It has recently been shown that changes in the TGF-β1 pathway may be involved in the pathogenesis of both chronic bronchitis and emphysema, which are the main clinical characteristics of COPD.8,9,16 It is known that TGF-β1 acts as a negative regulator of mucin production induced by bacterial infections, as shown by Jono and co-workers.16 These authors clearly demonstrated that infection with H influenzae induces a defensive response characterised by upregulation of mucins, and that TGF-β1 is crucial for attenuating this response after removal of the infectious agent. Indeed, these authors found that suppression of the TGF-β RII pathway resulted in mucin hyperproduction in vitro. In vivo it has been shown that a mouse knockout for Runx3, a transcription factor that mediates TGF-β signalling, develops mucus hypersecretion.21 Our finding of decreased TGF-β RII expression in smokers with COPD, most of whom had symptoms of chronic bronchitis, supports the view that changes in the TGF-β pathway may result in mucus hypersecretion in humans also.
Interestingly, when we examined the relationship between TGF-β RII expression and Reid’s index we observed a negative correlation, indicating that the lower the expression of this receptor, the larger the size of the bronchial gland. Moreover, when the mucus content in the bronchial glands was analysed, there was a trend for the correlation to be significant which suggests that the lower the expression of TGF-β RII, the higher the number of mucin positive acini. Taken together, these observations further support the protective role of the TGF-β pathway on mucus hypersecretion in smokers.
These findings are in line with the recent hypothesis that TGF-β1 might have a role in maintaining lung homeostasis in physiological conditions, an effect which may be lost in smokers who develop COPD.10,22 A similar role for TGF-β1 in maintaining tissue homeostasis has been proposed in other chronic diseases since decreased TGF-β RII activity has been observed in the early phases of tumorigenesis and atherogenesis.23,24
The concept that TGF-β1 may have a protective role in smokers is also supported by two recent genetic studies which showed that polymorphisms of the TGF-β1 gene associated with higher production of this growth factor were more frequent in healthy smokers than in smokers with COPD.10,11 Furthermore, studies on animal models strongly suggest that abnormalities in the activation and signalling of TGF-β1 are important in the pathogenesis of emphysema, and it has been shown that mice lacking activation of latent TGF-β1 develop severe pulmonary emphysema through alterations of macrophage metalloelastase MMP-12.8,9 A recent study has shown that similar mechanisms also occur in humans. Indeed, it has been shown that alveolar macrophages from patients with COPD have reduced TGF-β1 production which is paralleled by reduced production of TIMP-1.25 These findings suggest that impairment of TGF-β1 is associated with reduced antiproteolytic activity which may in turn result in emphysematous lesions. Similarly, we have recently shown that TGF-β RII expression in the alveolar walls of patients with severe emphysema is negatively correlated with T lymphocyte infiltrate which is thought to promote alveolar wall destruction.22
The intriguing concept which can be inferred from all these studies is that, by downregulating a single signalling pathway in the lung (TGF-β1), it is possible to promote the development of the two main clinical components of COPD—emphysema and chronic bronchitis. Nevertheless, it should be emphasised that the role of TGF-β1 in COPD is still controversial, with some studies reporting increased TGF-β1 signalling5–7 and others being unable to replicate these findings.26,27 However, it is difficult to compare our findings with those of others because, unlike previous studies, we focused specifically on the bronchial glands. It is conceivable that the expression of TGF-β1 and its receptors could be under the control of different factors in the different lung compartments.
Among its different activities, TGF-β1 is known to play a pivotal role in the resolution of immune responses12–14 and it can be hypothesised that decreased expression of TGF-β RII may result in a persistent inflammatory response. Indeed, in the present study we confirmed the prominent neutrophilia within the bronchial glands that we previously reported in smokers with COPD. However, when we examined the relationship between TGF-β RII expression and neutrophil numbers we found no significant correlation. These findings indicate that the absence of TGF-β signalling has a direct effect on gland hyperplasia and mucus hypersecretion, but not on neutrophilic infiltration within the glands.
Even if we favour the hypothesis that decreased TGF-β RII expression reflects decreased TGF-β signalling, an alternative explanation is that it would result from internalisation of the receptor due to excessive TGF-β binding. However, we think this is unlikely because it has been reported that internalisation of TGF-β RII occurs at an extremely slow rate28 and is independent of the presence of the ligand.29
One possible limitation of our study is that we were not able to include a group of healthy non-smoking subjects for comparison because most lung cancer patients are smokers. Nevertheless, when we measured the expression of TGF-β1 and its receptor in the three non-smoking subjects whose tissue was available, it was similar to that of smokers with normal lung function. Another possible limitation of our study is that potential confounding factors may interfere with the expression of TGF-β. For example, TGF-β is known to bind to different proteins in the extracellular matrix30 and this interaction may mask the molecule to the specific antibody. This interaction may raise concerns when considering the expression of the ligand, since TGF-β1 can be secreted outside the cell and interact with extracellular matrix proteins. However, we are confident that our finding of decreased TGF-β RII expression is valid because, unlike the ligand, the receptor is mostly present in the cell and is less likely to bind to components of the extracellular matrix. Finally, in the present study we examined only the expression of TGF-β1 and its type II receptor, while the TGF-β family includes many different ligands and receptors. We cannot therefore exclude the possibility that other members of the TGF-β family, in particular TGF-β2 which is known to be dysregulated in asthma, could also have a role in COPD.31
In conclusion, this study shows that there is decreased expression of TGF-β IIR in the bronchial glands of smokers with COPD which correlates with bronchial gland enlargement. These results suggest that TGF-β1 signalling may have a protective role in the development of mucus hypersecretion in COPD, a finding which is somewhat surprising. A similar protective role has recently been reported in other chronic disorders such as atherosclerosis and cancer where TGF-β1 was traditionally thought to have detrimental effects.
REFERENCES
Footnotes
-
Published Online First 14 October 2005
-
Supported by the Italian Ministry of University and Research and by the University of Padova (Grant CPDG 035929).
-
Competing interests: none declared.