Article Text

Abstract
Background: Urotensin II (UII) has been identified as a ligand for the orphan receptor GPR14 through which it elicits potent vasoconstriction in humans and non-human primates. The pulmonary vasculature is particularly sensitive; human UII (hUII) exhibits a potency 28 times that of endothelin (ET)-1 in isolated pulmonary arteries obtained from cynomolgus monkeys. However, hUII induced vasoconstriction in isolated human intralobar pulmonary arteries is variable, possibly as a result of location dependent differences in receptor density or because it is only uncovered by disease dependent endothelial dysfunction.
Methods: The vasoactivity of both hUII and gobi UII (gUII) in comparison with ET-1 and ET-3 was studied in isolated perfused lung preparations (n = 14) and isolated intralobar pulmonary arteries (n = 40, mean diameter 548 (27) μm) obtained from 17 men of mean (SE) age 67 (2) years and eight women of mean (SE) age 65 (3) years with a variety of vascular diseases.
Results: ET-1 (10 pM–100 nM) and ET-3 (10 pM–30 nM) elicited vasoconstriction in the lung preparations, inducing comparable increases in pulmonary arterial pressure of 24.8 (4.5) mm Hg and 14.5 (4.9) mm Hg, respectively, at 30 nM (p = 0.13). Similarly, ET-1 (10 pM–300 nM) and ET-3 (10 pM–100 nM) caused marked vasoconstriction in isolated pulmonary arteries, inducing maximal changes in tension of 4.36 (0.26) mN/mm and 1.54 (0.44) mN/mm, respectively, generating −logEC50 values of 7.67 (0.04) M and 8.08 (0.07) M, respectively (both p<0.05). However, neither hUII nor gUII (both 10 pM–1 μM) had any vasoactive effect in either preparation.
Conclusion: UII does not induce vasoconstriction in isolated human pulmonary arterial or lung preparations and is therefore unlikely to be involved in the control of pulmonary vascular tone.
- urotensin II
- endothelin
- pulmonary vasculature
- ET, endothelin
- gUII, goby urotensin II
- hUII, human urotensin II
- UII, urotensin II
Statistics from Altmetric.com
Urotensin II (UII) is a cyclic neurosecretory hormone initially identified in fish, being isolated and sequenced from goby Gillichthys mirabilis.1 Goby UII (gUII) is a 12-amino acid residue (AGTADCFWKYCV) which contains a cyclic hexapeptide (underlined) and is structurally similar to somatostatin-14.1 The human form of UII (hUII) has also been cloned2 and comprises 11 amino acids (ETPDCFWKYCV). The cyclic hexapeptide region is retained and appears to be conserved in all species from which UII has been cloned; it is proposed that this region confers the majority of its biological activity.3 hUII exhibits a high affinity for the GPR14 orphan G-protein coupled receptor which has now been termed the urotensin II receptor (UT receptor), and triggers contraction via activation of UT coupled phospholipase C (PLC) with the resultant generation of inositol triphosphate (IP3) and activation of protein kinases,4,5 and also through calcium mobilisation from the extracellular environment.3,5,6 The vasoconstrictive ability of hUII is lost in UT receptor knock-out mice,7 providing strong evidence that UII mediated contractions occurs via activation of this receptor. A series of efficacy studies in tissue from non-human primates showed that hUII was the most potent mammalian vasoconstrictor yet identified, eliciting responses an order of magnitude higher than endothelin-1 (ET-1).6 Indeed, in primates an infusion of only 300 pmol/kg hUII was sufficient to drastically increase total peripheral resistance and trigger a fatal circulatory collapse.6 UT receptor mRNA is extensively expressed in human cardiovascular tissues including the heart, vascular smooth muscle, and endothelial cells,6 and much interest has surrounded the vascular effects of UII and its potential pathological role in cardiovascular disease.
Despite exhibiting a marked vasoconstrictive effect, the consistency of the response to hUII appears to be highly variable and is dependent on both the species and the vascular bed.8 However, in non-human primates a consistent and potent constriction to hUII is reported in all arteries studied.6,8 These include thoracic and abdominal aortas and coronary, pulmonary, femoral, mesenteric, renal, carotid, basilar and internal mammary arteries.6,8 The pulmonary vasculature was particularly sensitive to hUII, exhibiting a potency 28 times that of ET-1.6 Such observations suggest that the vasoactivity of UII is restricted to—or is more prominent in—primates, and has led to speculation that hUII may be involved in the pathogenesis of human pulmonary vascular disease.
However, results gathered to date from human studies are inconclusive. Endothelial-denuded human coronary, mammary and radial arteries exhibit potent (albeit modest) vasoconstrictor responses to hUII,9 yet human pulmonary resistance arteries were non-responsive.10 However, the presence of l-NAME uncovered a variable vasoconstrictor response to hUII in 30% of the human pulmonary resistance arteries tested, and it was hypothesised that under normal conditions hUII may trigger release of nitric oxide which antagonises its contractile efficacy.10 hUII may therefore be of greater relevance in pathophysiological conditions in which endothelial dysfunction occurs, which is supported by the observation that pulmonary arteries harvested from pulmonary hypertensive rats exhibit an enhanced pressor response to hUII.10 The variability of the response to hUII in human pulmonary resistance arteries may also be due to disease specific differences in hUII receptor density.10 Unfortunately, the location of the lung tissue used in this previous study and the details of the patients from which it was obtained were unknown.9 No association could therefore be drawn between hUII vasoreactivity and the disease states or the vascular location of the vessels.
This study was undertaken to determine whether UII had any vasoconstrictive effect in isolated perfused intact human lungs or in isolated human pulmonary resistance arteries obtained from patients with various pulmonary and vascular diseases, and to compare the efficacy and potency of UII with that of ET-1 and endothelin-3 (ET-3).
METHODS
Tissue procurement
Full approval to undertake the study was obtained from the relevant local ethics committee. Lung samples were obtained from 17 men of mean (SE) age 67 (2) years and eight women of mean (SE) age 65 (3) years with lung cancer undergoing elective lobe or lung resection. Typically, the carcinoma occupied a minimal proportion of the lobe/lung volume. Patient details including their sex, age, medical history, and current medication are shown in tables 1 and 2. Details of the anaesthetic and analgesic regimen to which the patients were exposed during lung resection are also shown. Following resection of the lung sample, the pulmonary circulation was immediately flushed with an oxygenated Krebs bicarbonate solution to remove any residual blood or clots and the sample weighed. The tissue was then immediately transferred to the research laboratory in chilled Krebs bicarbonate solution and used either for perfusion studies (table 1) or as a source of isolated intrapulmonary arteries (table 2).
Details of lung samples used for isolated perfused human lung experiments and characteristics of patients from whom the tissue was obtained
Details of lung samples used for isolated human pulmonary artery experiments and characteristics of the patients from whom the tissue was obtained
Isolated perfused human lung model
The ex vivo saline perfused human lung system used in the present study is shown in fig 1. The lung sample was suspended in a polycarbonate collection reservoir (Sorin Biomedica, Quedgeley, Gloucester, UK) from a force transducer (Thames Side Maywood Instruments Ltd, Tilehurst, Berkshire, UK) to allow continuous measurement of lung weight. The pulmonary arterial and bronchial systems were cannulated using the largest cannulae possible, and the sample surrounded with plastic to prevent desiccation of the surface. The bronchial cannula was connected to a piston ventilator (Harvard Apparatus Ltd, Edenbridge, Kent, UK) to allow ventilation with room air. A respiration rate of 10 breaths/min and tidal volume of 100–300 ml were set according to the size and airway resistance of the lung sample and maintained for the duration of the experiment. The resultant mean airway pressure was 14.1 (3.4) mm Hg. The pulmonary artery cannula was then connected to the perfusion circuit which consisted of connective PVC tubing (Cobe Laboratories, Quedgeley, Gloucester, UK), which had been primed with 1 litre oxygenated Krebs bicarbonate solution to ensure that all air had been evacuated. The perfusate comprised Krebs bicarbonate solution aerated with 95% O2/5% CO2 and was circulated to the pulmonary arteries from a second polycarbonate perfusate reservoir (Baxter Healthcare, Compton, Berkshire, UK) via a peristaltic pump (Watson-Marlow, Falmouth, Cornwall, UK). The perfusate temperature and pH were continuously recorded using in line temperature (Terumo UK, Knowsley, Merseyside, UK) and pH (Philips Medical Systems, Leeds, West Yorkshire, UK) probes. The temperature was maintained at 36–38°C by a heat exchanger (Sorin Biomedica) and the pH at 7.4 via a pH controller (Medical Physics, Hull Royal Infirmary, Hull, UK) which regulated the flow of CO2 into the perfusate. Pulmonary venous drainage flowed freely into the collection reservoir before being returned to the perfusate reservoir for re-circulation. Perfusion pressure was continually monitored via a pressure transducer (Datex-Ohmeda Ltd, Hatfield, Hertfordshire, UK) and maintained at 18.9 (4.6) mm Hg by regulating the perfusion flow rate. This was determined for each sample on the basis of the size and vascular resistance of the sample, and ranged between 100 and 500 ml/min. Regular “arterial” and “venous” perfusate samples were collected for analysis of pH, Pco2, Po2, Na+, K+, iCa++, HCO3, HCO3 std, Tco2, and base excess in a blood gas analyser (Mallinckrodt, Bicester, Oxfordshire, UK). “Arterial” samples were obtained from the perfusion line just proximal to the arterial cannulae. “Venous” samples were obtained by collecting a small volume of the effluent from the sample in the collection reservoir. Analysis of perfusate samples confirmed that this system efficiently replaced exhaled CO2 and returned the pH, Pco2, HCO3 and Tco2 of the perfusate to within normal physiological parameters before it was re-circulated to the lung sample. The mean time from removal of the lung sample from the patients and starting the experiment was 43 (6) minutes.
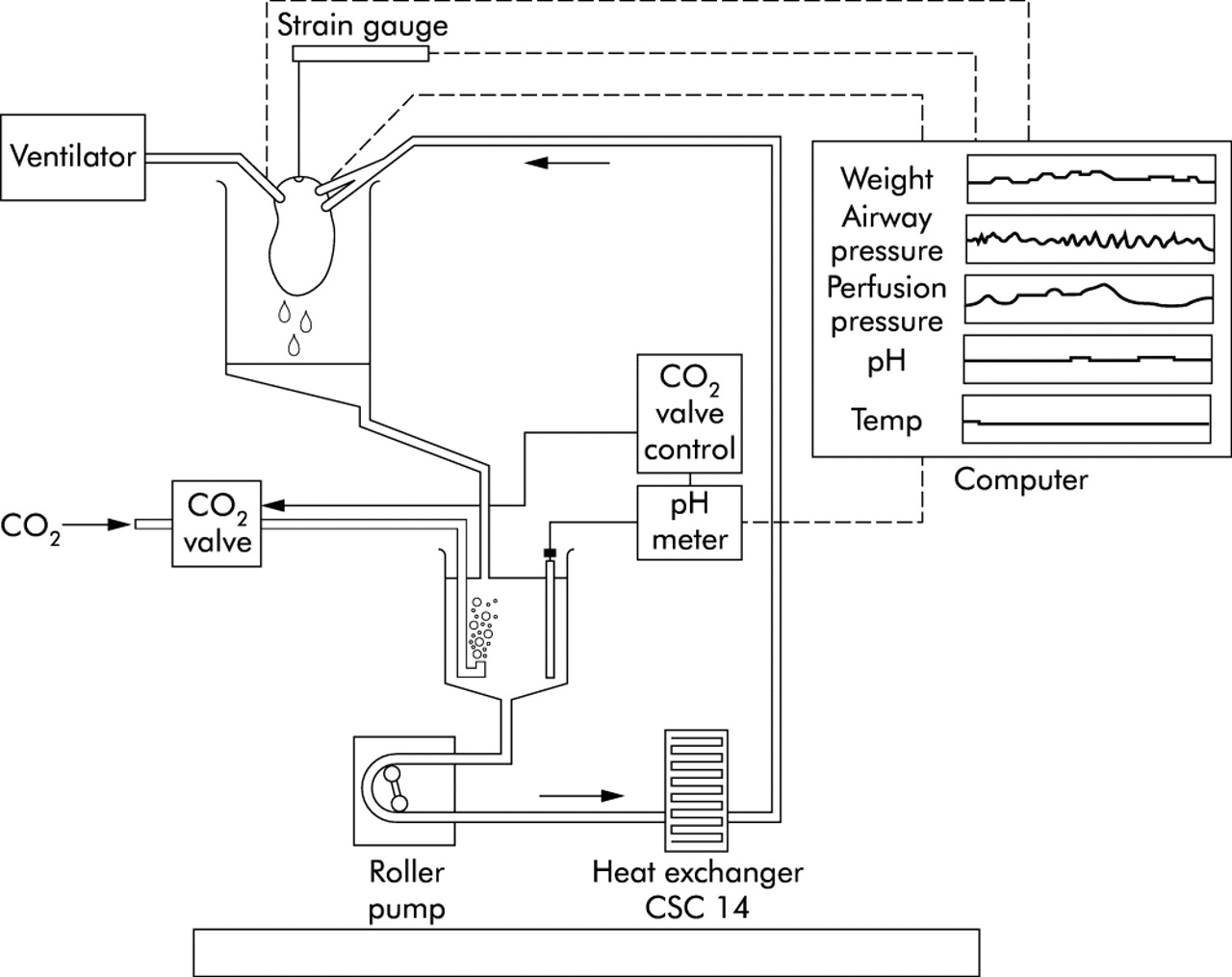
Ex vivo saline perfused human lung system used in the present study. Following resection the pulmonary arterial and bronchial system were cannulated and the lung sample wrapped in a protective clear plastic film. The lung sample was then suspended from the force transducer (strain gauge) inside the collection reservoir. The bronchial cannula was connected to the piston ventilator to allow ventilation with room air. The pulmonary arterial cannulae were connected to the perfusion circuit which consisted of connective PVC tubing. Krebs bicarbonate solution was placed in the perfusate reservoir and circulated to the pulmonary arterial cannulae via a peristaltic roller pump. The pH of the perfusate reservoir was monitored via a pH meter which activated the supply of additional CO2 into the perfusate via the CO2 valve control when the pH exceeded 7.4. The perfusate temperature was maintained at 36–38°C by a heat exchanger and was continually monitored by the temperature probe. Pulmonary venous drainage flowed freely into the collection reservoir and was returned to the perfusate reservoir for re-circulation. Perfusion pressure was continually monitored via a pressure transducer located proximal to the pulmonary arterial cannulae.
Once stable baseline airway and perfusion pressures were obtained, the pharmacological effects of ET-1, ET-3, or hU-II were assessed by direct addition of each peptide into the perfusate reservoir. Cumulative concentration-response curves were constructed to either ET-1 (0.01–100 nM, n = 8) or ET-3 (0.01–30 nM, n = 7) which, in some cases, was preceded by exposure to hUII (0.01 nM–100 nM, n = 3; table 1), and the change in perfusion pressure recorded. The viability of each lung sample was confirmed by a significant increase in perfusion pressure in response to either ET-1 or ET-3. In the three lung samples initially exposed to hUII the perfusate was completely exchanged by flushing the system with 3 litres fresh Kreb’s solution over a 45 minute period. This perfusate was then discarded and fresh Kreb’s solution used to perfuse each sample before exposure to either ET-1 (0.01–100 nM) or ET-3 (0.01–30 nM). Agonist concentration-response curves were repeated after a minimum of 45 minutes and when the baseline pressures returned to normal.
Isolated human pulmonary arteries
Intralobar pulmonary arteries (n = 40, mean (SE) internal diameter 548 (27) μm) were carefully dissected from healthy areas of lung sections obtained from male (n = 8) and female patients (n = 2) as shown in table 2. Vessels were then mounted on two 40 μm stainless steel wires between the jaws of an automated small vessel wire myograph (Cambustion Ltd, Cambridge, UK) as previously described.11 Extreme care was taken to avoid damage to either the smooth muscle or endothelial cell layers. The myograph bath contained 5 ml physiological saline solution (PSS) warmed to 37°C and bubbled with 95% O2/5% CO2 to maintain a pH of 7.4. Length-tension curves were plotted for each vessel before loading to a tension equivalent to a transmural pressure of 17.5 mm Hg, the normal in vivo resting tension. The vessels were then left to equilibrate for 1 hour before exposure to 80 mM potassium chloride (KCl). The contraction was allowed to proceed until a maximal tension was obtained, after which the vessels were washed by changing the PSS twice and left to re-equilibrate back to their original basal tension. The vessels were then re-exposed to 80 mM KCl to confirm that the vessels were responding consistently to a given concentration of agonist, then washed by changing the PSS twice. Any vessel which had a mean contraction to 80 mM KCl of <1 mN/mm was assumed to be damaged during the mounting procedure and was excluded from the study. Four vessels were excluded on this basis. Once a stable baseline tension had been re-established, the endothelial integrity of each vessel was confirmed by significant vasodilatation to acetylcholine (ACh, 1 μM) following preconstriction with prostaglandin F2α (PGF2α, 100 μM). The vessels were then washed and, once the original basal tension had been regained, exposed to cumulative doses of either gUII (0.01 nM–1 μM; n = 10), hUII (0.01 nM–1 μM; n = 10), ET-1 (0.01–300 nM; n = 8), or ET-3 (0.01–100 nM; n = 8). At the end of the protocol vessels were washed by changing the PSS in the myograph bath twice, then re-exposed to 80 mM KCl to confirm viability.
Materials and reagents
Krebs bicarbonate solution consisted of 113.8 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 25 mM NaHCO3, 1.2 mM KH2PO4, 11.4 mM glucose, and 2.4 mM CaCl2 dissolved in distilled water. PSS consisted of 120 mM NaCl, 4.7 mM KCl, 1.17 mM MgSO4, 25 mM NaHCO3, 1.18 mM KH2PO4, 5.5 mM glucose, 2.5 mM CaCl2, and 26.9 μM EDTA dissolved in distilled water. ET-1 was a gift from AstraZeneca, Macclesfied, Cheshire, UK. ET-3 was obtained from Cambridge Research Biochemicals, Billingham, Cleveland, UK, and human UII was obtained from Peninsula Laboratories, St. Helens, Merseyside, UK. All other chemicals were obtained from Sigma, Poole, Dorset, UK. All reagents were dissolved in distilled water.
Statistical analysis
All values are expressed as mean (SE) and were analysed by ANOVA, Student’s unpaired t test, Mann-Whitney U test, or Kruskal-Wallis test where appropriate. −logEC50 values were calculated by hand from individual concentration-response curves which had reached their maximal response following exposure to the highest concentration (this was not possible for one of the isolated perfused lung models). Significance was assumed at p values of <0.05.
RESULTS
Isolated perfused human lung model
No significant differences were seen in the characteristics of the patients or lung tissue used in the study. The mean age of the patients from whom lung samples were obtained for exposure to ET-1, ET-3, or hUII was 67 (2) years (n = 8), 67 (3) years (n = 7), and 71 (3) years (n = 3), respectively (p = 0.64, ANOVA). The mean basal pulmonary artery pressure of lung samples exposed to ET-1, ET-3, or hUII was 18.9 (4.7) mm Hg, 17.1 (5.2) mm Hg, and 23.3 (4.5) mm Hg, respectively (p = 0.80, ANOVA). The mean basal airway pressure of lung samples exposed to ET-1, ET-3, or hUII was 11.8 (2.2) mm Hg, 16.3 (3.3) mm Hg, and 14.0 (3.6) mm Hg, respectively (p = 0.62, ANOVA). The mean weight of the lung samples exposed to ET-1, ET-3, or hUII was 392 (57) g, 508 (118) g, and 440 (82) g, respectively (p = 0.71, ANOVA).
Both ET-1 and ET-3 elicited vasoconstriction in the isolated perfused human lung preparations, observed as a marked increase in the pulmonary arterial pressure. Maximal vasoconstriction to ET-1 occurred at a concentration of 100 nM; Emax = 28.1 (4.4) mm Hg with a −logEC50 of 8.08 (0.15) (fig 2). Prohibitive peptide costs prevented the study of ET-3 at concentrations higher than 30 nM, but the maximal response at this concentration was 14.5 (4.9) mm Hg which was not significantly different from the response to ET-1 at this concentration: 24.8 (4.5) mm Hg (p = 0.14, Student’s unpaired t test, fig 2). In contrast, hUII had no effect on pulmonary artery pressure (fig 2).

Concentration-response curves in isolated perfused human lung preparations for ET-1 (□, n = 8), ET-3 (▪, n = 7) and hUII (•, n = 3).
Isolated human pulmonary arteries
No significant differences were seen in either the patient or vessel characteristics used in this study. The mean age of the patients from whom pulmonary arteries were obtained for exposure to ET-1, ET-3, hUII, or gUII was 71 (3) years (n = 4), 67 (3) years (n = 4), 67 (3) years (n = 5), and 66 (3) years (n = 5), respectively (p = 0.96, ANOVA). The mean internal diameter of the pulmonary arteries exposed to ET-1, ET-3, hUII, or gUII was 504 (42) μm (n = 10), 679 (52) μm (n = 10), 483 (47) μm (n = 8), and 517 (52) μm (n = 10), respectively (p = 0.07, ANOVA). The mean contractile response to the two initial exposures to 80 mM KCl before addition of ET-1, ET-3, hUII, or gUII was 4.00 (0.63) mN/mm, 4.24 (1.02) mN/mm, 3.91 (0.73) mN/mm, and 3.46 (0.51) mN/mm, respectively (p = 0.80, ANOVA). Statistically similar responses were produced to the final addition of 80 mM KCl undertaken following exposure to ET-1, ET-3, hUII, or gUII: 2.97 (0.83) mN/mm (p = 0.05, ANOVA), 3.72 (0.98) mN/mm (p = 0.71, ANOVA), 4.04 (0.74) mN/mm (p = 0.92, ANOVA), and 3.66 (0.47) mN/mm (p = 0.87, ANOVA), respectively. The mean vasodilatory response to 1 μM ACh (following preconstriction with 100 μM PGF2α) of the pulmonary arteries exposed to ET-1, ET-3, hUII, or gUII was −29.7 (14.9)%, −31.2 (15.3)%, −27.6 (12.9)%, and −30.0 (16.2)%, respectively (p = 0.88, Kruskal-Wallis test).
Both ET-1 and ET-3 elicited marked vasoconstriction in isolated human pulmonary arteries (fig 3). Maximal vasoconstriction to both peptides occurred at a concentration of 100 nM; Emax to ET-1 was 4.36 (0.26) mN/mm with an EC50 of 7.67 (0.04), while Emax to ET-3 was 1.54 (0.44) mN/mm (p<0.05, Student’s unpaired t test) producing an EC50 of 8.08 (0.07) (p<0.05, Student’s unpaired t test). In contrast, neither hUII nor gUII had any vasoconstrictive efficacy in isolated human pulmonary arteries, even at concentrations of 1 μM (fig 3).

{kind=link}
{kind=link}
{kind=link}
Concentration-response curves in isolated human pulmonary arteries to ET-1 (□, n = 8), ET-3 (▪, n = 8), hUII (•, n = 10), and gUII (○, n = 10). *p<0.05 (Student’s unpaired t test).
DISCUSSION
This is the first study to assess the vasoconstrictive efficacy of UII in isolated perfused human lungs, and the first to attempt to link the consistency of UII mediated vasoconstriction to disease state and endothelial function in isolated human pulmonary arteries. The main finding of the study was that, while ET-1 and ET-3 produced marked and potent vasoconstriction in these preparations, neither gUII nor hUII had any demonstrable vasoconstrictive activity.
A great deal of interest has surrounded the vascular actions of UII following the discovery that it was able to elicit potent vasoconstriction following activation of the GPR14 orphan receptor.6 GPR14 (now termed UT receptor) is a 389-residue transmembrane protein similar in structure to the somatostatin receptor sst4.12 Binding of UII to UT is reported to elicit calcium mobilisation,6 although the precise signalling cascade which precedes this increase in intracellular calcium remains unclear, with activation of voltage gated calcium channels,5,13 phospholipase C,4,5 protein kinase C,5,14 tyrosine kinase,5 and Rho-kinase5,15 all being proposed to be involved.
Ames et al6 were the first to report that hUII elicited vasoconstriction in non-human primates with a potency greater than ET-1. Thoracic and abdominal aortas and coronary, pulmonary, mesenteric, renal, carotid, basilar, and internal mammary arteries were all reported to be sensitive to the vasoconstrictive action of hUII.6 Endothelial-denuded human coronary, mammary and radial arteries are also reported to exhibit potent (albeit modest) vasoconstrictor responses to hUII, as are human saphenous and umbilical veins.9 This in vitro activity is mirrored by in vivo studies; infusion of hUII results in a dose dependent reduction in forearm blood flow in healthy male volunteers, consistent with a peripheral contractile response,16 while in adult male cynomolgus monkeys infusion of UII at doses of 300 pmol/kg was sufficient to drastically increase peripheral resistance and trigger fatal circulatory collapse.6 However, this marked vasoconstrictive efficacy would not appear to be translated into animal preparations. Douglas et al8 conducted a thorough screening study of the vasoconstrictive efficacy of hUII in a vast array of vascular preparations from a variety of mammalian species (rat, mouse, pig, dog, and cynomolgus monkey). While this report supported that of Ames et al6 in that hUII was able to contract thoracic and abdominal aortas and coronary, pulmonary, mesenteric, renal, carotid, basilar and internal mammary arteries of primates, of the other animal preparations studied only rat thoracic aortas and canine coronary arteries were reported to exhibit a similar sensitivity to hUII.8 Restriction of the vasoconstrictive efficacy to the mammalian thoracic aorta has also previously been reported for gUII.3,13,17 The efficacy of UII would therefore appear to be restricted to vascular tissue obtained from humans and non-human primates, a species specificity which may have direct consequences for human disease.
The primate studies indicated that pulmonary arteries were particularly sensitive to the vasoconstrictive effects of hUII, being 28 times more potent than ET-1.6 Such findings prompted the suggestion that UII may have a potential role in pulmonary vascular disease and conditions characterised by excessive pulmonary vasoconstriction such as pulmonary hypertension.
The first study to investigate the vasoactive properties of hUII in the pulmonary vasculature was that of MacLean et al10 who used both intralobar human pulmonary arteries and intralobar and extralobar pulmonary arteries obtained from normal and pulmonary hypertensive rats. hUII was a potent and efficacious constrictor of extralobar rat pulmonary arteries. The efficacy of hUII was increased further by pretreatment with the nitric oxide synthase inhibitor l-NAME, by the increase of basal pulmonary arterial tone via exposure to ET-1, and in vessels harvested from animals exposed to chronic hypoxia for 2 weeks and confirmed to be pulmonary hypertensive.10 Endothelial denudation resulted in an increase of about 2.5 times in the potency of hUII.10 However, hUII had no efficacy in intralobar rat pulmonary arteries or in human pulmonary resistance arteries,10 observations consistent with our findings. Pretreatment with l-NAME uncovered a vasoconstrictor response to hUII in three of the 10 human pulmonary resistance arteries tested, although this varied enormously between vessels (14%, 38%, and 220% of the response to 50 mM KCl) and was slow in onset.10 This variable response was hypothesised to be due to differences in hUII receptor density within the vasculature, or may constitute an activity which is only uncovered following endothelial dysfunction arising as a consequence of the disease state. Unfortunately, the human vessels used in this previous study were obtained from unknown regions of lung and the patient’s disease state was not reported.
The results of the present study do not support this hypothesis. As can be seen from tables 1 and 2, the lung samples used either for whole lobe/lung perfusion or as a source of intralobar pulmonary arteries were obtained from patients with a broad spectrum of conditions including hypertension, coronary heart disease, peripheral vascular disease, and diabetes where endothelial dysfunction is well recognised. Furthermore, despite the fact that ACh evoked a mean relaxation of approximately 30% in the isolated pulmonary arteries used in our study, the response was markedly varied (ranging from +10% to −60%). Consequently, both vessels without a functional endothelial cell layer and vessels with a diverse range of endothelial activity were included. Since neither hUII nor gUII evoked any demonstrable vasoconstriction in either these isolated intralobar pulmonary arteries with different endothelial status or in isolated perfused whole lungs, there is no evidence to suggest that UII induces pulmonary arterial contraction which is dependent upon vascular location, disease status, or endothelial function. The possibility of degradation of the hUII peptide was excluded by parallel studies which showed that exposure of isolated rat thoracic aorta (n = 4) to the same concentration of hUII as that used in the present study (using the same solutions) induced a maximal vasoconstriction of 0.52 (0.11) mN/mm (equivalent to approximately 20% of the reference contraction to KCl, consistent with the literature for this vascular bed7). Similarly, the viability of the preparations was confirmed by significant vasoconstrictor responses to either ET-1 or ET-3 (isolated lungs) or KCl (isolated arteries) being observed following exposure to UII. An alternative explanation for the lack of efficacy of UII may arise from the fact that the experiments were undertaken in tissue obtained solely from patients with chronic disease. Although, as discussed previously, such conditions are postulated to be necessary to uncover the vasoconstrictive activity of UII, this remains a possibility. However, we have previously reported that hUII and gUII are also ineffective in inducing vasoconstriction in isolated rat intralobar pulmonary arteries.18 Although this is a cross species comparison, it does provide some evidence that the presence of underlying disease in the present study is not masking vasoconstrictive activity of UII. It would appear therefore that UII does not act as a vasoconstrictor in the pulmonary circulation.
In contrast to UII, ET-1 induced marked pulmonary vasoconstriction. The observed responses to ET-1 were similar in both isolated pulmonary arteries and isolated perfused lungs, and were also similar to those previously reported in rat pulmonary arteries by our group,19 consistent with activity at the ETA receptor. This also shows that the tissue used in the present study was reacting in a similar manner to non-diseased tissue. However, significantly lower responses to ET-3 than to ET-1 were observed at 100 nM in the isolated pulmonary arteries (fig 3). The most likely explanation for this discrepancy is that this response is also mediated via ETA receptors, since ET-3 is widely recognised to exhibit a lower agonist potency for this receptor than ET-1.20 The vasoconstrictive action of ET-1 and ET-3 in isolated perfused lung preparations is also likely to be ETA mediated since comparable responses to ET-1 and ET-3 were observed in both preparations. Indeed, the ET-1 and ET-3 responses are almost super-impossible, which strongly suggests that the same receptors mediate the response in both isolated pulmonary arteries and isolated perfused lungs. However, because statistically similar responses are observed up to 30 nM in isolated perfused lung preparations, it is possible that contractile ETB2 receptors may be involved. However, there is no evidence for the involvement of ETB2 receptors in the response in isolated pulmonary arteries and these receptors are not expressed in all tissue.
Other investigators have also found a lack of contractile efficacy for hUII in isolated human arteries. Hillier et al21 reported that isolated subcutaneous resistance arteries and internal mammary arteries obtained from both healthy volunteers and patients with vascular disease are also resistant to hUII. Furthermore, Behm et al7 reported that the UT receptor knock-out mouse does not exhibit abnormal systemic haemodynamics, blood pressure or heart rate, does not show any echocardiographic abnormalities, and affected animals have no changes in major organ weight. Similarly, vascular reactivity to a wide variety of other spasmogens is unaffected.7 Such observations also preclude a disease dependent vasoconstrictive action and suggest that hUII has little involvement in human cardiovascular physiology or pathophysiology. Also, in the study of Maguire et al,9 although hUII was reported to contract endothelium-denuded human coronary, mammary and radial arteries with a potency about 50 times that of ET-1, the responses only constituted 15–19% of the contraction observed to 100 mM KCl and were not observed in all vessels studied. Similarly, Douglas et al8 have reported modest hUII induced contraction in isolated porcine intralobar pulmonary arteries, but the responses generated were only observed in two of the four vessels tested.
An alternative explanation for the lack of response to hUII in the present study may be that it is acting as a vasodilator rather than a vasoconstrictor. Stirrat et al22 have recently shown that hUII evokes marked vasodilatation in isolated human pulmonary resistance arteries preconstricted with ET-1 although, in the study by MacLean et al,10 pulmonary arteries preconstricted with ET-1 exhibited only pressor responses to hUII. A similar vasodilatory activity is also reported in human systemic resistance arteries22 and in rat coronary23,24 and mesenteric arteries.24 However, both the isolated lungs and isolated arteries in the present study were investigated at the in vivo pressure and were therefore equally receptive to both vasoconstrictor and vasodilator stimuli. Despite this, we were still unable to observe any vasodilatory activity of hUII or gUII in either of these preparations.
In summary, despite hUII having been previously reported to be an extremely potent vasoconstrictor in human and primate preparations, our results clearly show that this phenomenon does not extend to the human pulmonary vasculature. Neither hUII nor gUII had any demonstrable vasoactivity in either isolated perfused human lungs or in isolated human intralobar pulmonary arteries. UII is therefore unlikely to be involved in either the normal or pathogenic regulation of human pulmonary vascular tone.