Article Text

Abstract
The case history of a patient admitted to the ICU with severe hypoxic respiratory failure later diagnosed as Wegener’s granulomatosis is presented. The diagnosis and management of patients with suspected pulmonary vasculitis is discussed.
- critical care
- pulmonary vasculitis
Statistics from Altmetric.com
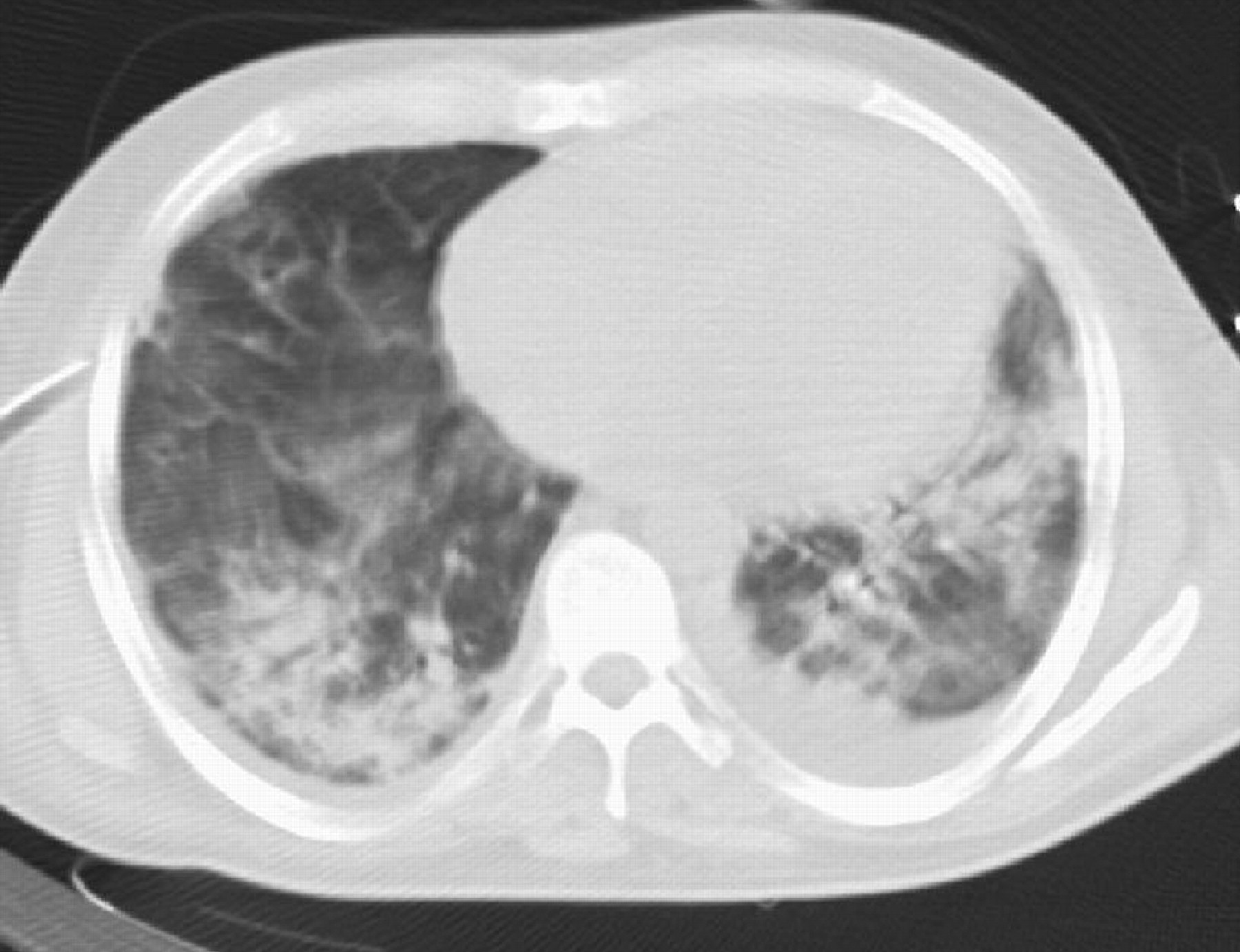
A 28 year old man was admitted from a local hospital to the intensive care unit (ICU) at Hammersmith Hospital via the operating theatre. He had presented with a 3 month history of nasal stuffiness, epistaxis and haemoptysis, and had recently noticed increasing shortness of breath. He had lost 6 kg in weight and reported night sweats. His initial chest radiograph showed diffuse peripheral shadowing and a subsequent computed tomographic (CT) scan confirmed the presence of multiple infiltrative lesions (fig 1). Bronchoscopy showed evidence of recent diffuse haemorrhage; bronchoalveolar lavage cytology revealed an eosinophilia but was negative for acid fast bacilli, legionella, and fungi. Over the next few days he deteriorated, requiring endotracheal intubation and ventilation. He was transferred for an open lung biopsy. Lung histology was non-specific showing diffuse alveolar haemorrhage with fibrin deposition, fibroblastic proliferation, and a predominantly neutrophilic inflammatory infiltrate. Following the lung biopsy the patient was admitted to the ICU with severe hypoxaemic respiratory failure.
{kind=link}
CT scan of a ventilated patient with pulmonary haemorrhage. Scattered dense airspace shadowing can be seen with some background ground glass shadowing. Small effusions are also visible. Courtesy of Dr Jeremy Levy.
The patient had no significant past medical history. He was born in India and raised in the UK; his last visit abroad was to India 2 years before admission. He was a sales manager for a building company and did not smoke. On admission to ICU he was pyrexial at 38°C. He was paralysed and ventilated requiring 95% oxygen. There were no rashes, eyes and joints were normal, but he was noted to have a slight crusting of his nares. The blood pressure was 114/54 mm Hg supported by norepinephrine and dopexamine infusions. Clinical biochemistry results were: C reactive protein 99 mg/l, Na 135 mmol/l, K 4.3 mmol/l, urea 6.1 mmol/l, creatinine 52 μmol/l, albumin 16 g/l, ionised calcium 1.88 mmol/l, bilirubin 8 μmol/l, alkaline phosphatase 134 U/l (normal range 30–130), and alanine aminotransferase 76 U/l (0–31). The blood film showed a leucoerythroblastic picture; the counts were haemoglobin 7.5 g/dl, mean cell volume 89 fl, white cell count 15 × 109/l (neutrophil leucocytosis), platelets 324 × 109/l. Urine microscopy showed multiple red cell casts. Urgent ANCA (antineutrophil cytoplasmic antibodies) immunofluorescence was strongly positive for C-ANCA; anti-PR3 ELISA was also strongly positive at 89% (0–15%). This confirmed the clinical diagnosis of Wegener’s granulomatosis and treatment was initiated with prednisolone and cyclophosphamide. In view of the severity of his lung haemorrhage he underwent five episodes of plasma exchange. His renal function deteriorated over 6 days and he required continuous renal replacement therapy for fluid balance and metabolic control. The aetiology of the renal failure was multifactorial with sepsis and nephritis contributing. As he was receiving heavy immunosuppression, he was given anti-infective prophylaxis with co-trimoxazole, fluconazole, and isoniazid. In order to minimise further lung haemorrhage, his intravascular volume was kept as low as possible by tightly controlling fluid balance. He sustained one episode of line sepsis treated with vancomycin. He required high concentrations of oxygen (Fio2 >0.5) for 10 days and then his condition gradually improved; a tracheostomy was performed and his trachea was ultimately decannulated 15 days after ICU admission. His renal function recovered and he was discharged from hospital 46 days after admission with a creatinine level of 86 μmol/l. His pulmonary function was clinically good and was not assessed formally.
DISCUSSION
Presentation and diagnosis
This case illustrates the importance of the aggressive pursuit and treatment of the cause of acute respiratory failure. The history in this case provided important clues, but in some cases of acute severe respiratory failure a clinical picture of hypoxaemic respiratory failure with non-specific changes on the chest radiograph is all that is available on presentation to the ICU. The initial differential diagnosis is wide and includes most obviously infective agents, but also interstitial lung disease and vasculitis. High resolution CT scanning can be helpful but, unless it is performed in held static inspiration with arms out of the field, can be misleading. Serological tests are extremely valuable but may not be rapidly available.
Systemic vasculitis is characteristically a multifocal disease with a variety of clinical manifestations according to the size of the blood vessel involved and the organs affected. It is classified according to the size of the smallest vessel involved.1 Alveolar haemorrhage results from small vessel vasculitides involving the lungs; these include Wegener’s granulomatosis, microscopic polyangiitis and, rarely, Churg-Strauss syndrome. They can present at any age but occur most commonly in patients in their 50s and 60s and there is a slight male predominance.2,3 There are reports of vasculitis occurring in families,4–7 but most cases arise sporadically and, unlike most autoimmune diseases, so far no consistent HLA associations have been identified,8 although there are associations with certain complement alleles and alleles of α1-antitrypsin.9–12 Environmental factors such as silica exposure,13–15 drugs,3,16 and infections17–19 may play a part; however, although these may be important in elucidating the pathogenesis of vasculitis, in practice most cases have no obvious genetic or environmental precipitant. As can be seen from this case presentation, prompt diagnosis of vasculitis is vital, not only to instigate appropriate treatment but also to avoid unnecessary invasive diagnostic procedures. In retrospect, the previously arranged open lung biopsy in this case was probably not required and was precipitated by the deteriorating state of the patient and the need to secure a diagnosis. The ANCA result arrived on the ITU at the same time as the patient returned from theatre.
Small vessel vasculitis in the lung involves destruction of arterioles, capillaries and venules by an infiltration of activated neutrophils. This results in interstitial oedema and diffuse alveolar haemorrhage. Repeated episodes of lung haemorrhage may result in pulmonary fibrosis.20 Haemoptysis is a common presenting feature, although it can be late or even absent even in the face of significant alveolar bleeding.21 Other symptoms include dyspnoea, fever, and weight loss. Patients are anaemic, hypoxaemic, and have diffuse infiltrative shadowing on the chest radiograph. Alveolar haemorrhage can sometimes be confirmed on bronchoscopy, with haemosiderin-laden macrophages seen on cytological examination of lavage fluid. However, patients may be too hypoxic to undergo this procedure. Carbon monoxide transfer coefficient corrected for lung volume (Kco) will be raised. In the ITU patients are often ventilated, and methods of measuring Kco in ventilated patients have been reported and validated, although they are often only available in specialist centres where operators are familiar with their interpretation.22,23 A CT scan of the chest is often unhelpful in determining the aetiology of alveolar haemorrhage, although it may reveal cavitating lesions typical of granulomatous infiltration. Lung biopsies frequently demonstrate necrotic tissue or show non-specific inflammation and haemorrhage, particularly if obtained via the transbronchial route, but the yield can be improved by open lung biopsy. If there is evidence of renal involvement a renal biopsy is more likely to be diagnostic and will usually show a focal necrotising glomerulonephritis.
Patients with vasculitis may present with isolated pulmonary haemorrhage, but usually there is also systemic involvement. Most patients with Wegener’s granulomatosis have granulomatous lesions in the upper respiratory tract which result in chronic sinusitis, epistaxis, chronic otitis media, and deafness. Involvement of the trachea can present with life threatening stridor. Granulomas are also found in many sites outside the respiratory system including the kidney, central nervous system, prostate, parotid and orbit.3 Patients with microscopic polyangiitis and Wegener’s granulomatosis also present with systemic symptoms secondary to the small vessel vasculitis. Urine abnormalities include microscopic haematuria and proteinuria, with red cell casts on microscopy. The serum creatinine may initially be in the normal range, but acute renal failure tends to develop quite rapidly. Renal and respiratory involvement both have a significant effect on mortality.3,24,25 Other organs in which vasculitis commonly occurs are skin, joints, muscles, gastrointestinal system, peripheral and central nervous system, and the eye.
Routine blood counts often show a normocytic or microcytic anaemia with a neutrophil leucocytosis, although there can also be eosinophilia (especially in Churg-Strauss syndrome) and thrombocythaemia. Thrombocytopenia suggests alternative causes of lung haemorrhage such as primary haematological abnormalities or systemic lupus erythematosus, in which case there will often be accompanying low complement levels and positive antinuclear antibodies. The erythrocyte sedimentation rate and C reactive protein level will both be raised. Biochemistry will often show a low serum albumin, raised alkaline phosphatase, and raised urea and creatinine. These results may point to a diagnosis of vasculitis but are relatively non-specific. The most important serological tests in the diagnosis of vasculitis are antineutrophil cytoplasmic antibodies (ANCA). ANCA were first described in 198226 and subsequently was found to be associated with both Wegener’s granulomatosis and microscopic polyangiitis. There are two main patterns of ANCA—cytoplasmic (C) and perinuclear (P)—defined by their appearance on indirect immunofluorescence using ethanol fixed neutrophils. The main target antigen of C-ANCA is proteinase 3 (PR3).27–30 P-ANCA have a wider range of specificities but, in systemic vasculitis, they are usually specific for myeloperoxidase (MPO).31 Most patients with Wegener’s granulomatosis have anti-PR3 specific ANCA, patients with Churg-Strauss syndrome and polyarteritis nodosa have anti-MPO ANCA, and patients with microscopic polyangiitis have either anti-PR3 or anti-MPO ANCA. If immunofluorescence is used alone, then C-ANCA is more specific than p-ANCA for vasculitis since perinuclear staining is not only produced by anti-myeloperoxidase antibodies but also by antinuclear antibodies and antibodies to neutrophil enzymes not associated with vasculitis. All serum that is ANCA positive should therefore be tested in PR3 and MPO ELISA as this greatly increases their specificity.32 In a large European trial for the standardisation of ANCA assays, the specificity of immunofluorescence alone was found to be 97% for C-ANCA and 81% for a P-ANCA pattern. The combination of C-ANCA with anti-PR3 and P-ANCA with anti-MPO both had a specificity of 99%.33 However, a negative ANCA assay does not exclude the possibility of vasculitis as there are a few cases of small vessel vasculitides that are ANCA negative.34,35 In these cases a tissue diagnosis should be sought.
Management
Systemic vasculitis should be treated with cyclophosphamide (we use 3 mg/kg/day, reduced in the elderly to 2 mg/kg/day) combined with prednisolone. Historically, cyclophosphamide was continued for at least a year after remission.36,37 However, in view of the side effect profile of cyclophosphamide, many clinicians now substitute azathioprine in stable patients at 3 months. The European Vasculitis Study Group has set up a series of multicentre randomised controlled trials to investigate the roles of different immunosuppressive regimes in vasculitis. The first of these trials to be completed has recently confirmed that azathioprine is as effective as cyclophosphamide for maintenance of remission after 3 months,38 More recently mycophenylate mofetil, levamisole, and TNF directed treatment have also been used.39 In patients with fulminant disease such as dialysis dependent renal failure and alveolar haemorrhage, there may be an additional benefit from plasma exchange or intravenous methylprednisolone40,41 and the results of the current European trials should clarify this. In patients who fail to respond to this treatment or who cannot tolerate cyclophosphamide due to marrow suppression, there may also be a role for immunoabsorption,42,43 pooled immunoglobulin,44,45 or monoclonal antibodies,46 although so far much of the evidence is anecdotal. Lung haemorrhage is a major cause of early mortality and aggressive supportive therapy is important. Patients should be kept relatively intravascularly depleted until the lung haemorrhage is controlled, and clearly exacerbating pulmonary oedema must be avoided. Although acute renal failure may be exacerbated by this regimen, this is of secondary importance as the patient can be supported with renal replacement therapy and acute tubular necrosis caused by initial volume depletion is likely to recover in the long term. However, care must be taken to avoid hypoperfusion of the splanchnic circulation with consequent gut and liver dysfunction that may result in multiple organ failure.
There is little evidence on which to base ventilatory strategies for adults. A rational approach is to limit excessive tidal volume excursions or pressure changes which are likely to damage further the fragile vasculature and exacerbate haemorrhage. Modest gas exchange targets should therefore be set. However, profound hypoxaemia and hypercapnia should be avoided as this may exacerbate pulmonary hypertension. A high inspired oxygen fraction may be harmful on theoretical grounds concerning oxidant stress, but a compromise needs to be found until the patient responds to specific treatment. Extracorporeal membrane oxygenation has been used successfully in a small number of cases47 but cannot currently be recommended.
In summary, patients admitted to the ICU with suspected pulmonary vasculitis have a high mortality of around 25–50%, depending upon aetiology. These patients should be discussed early with a centre which has specialist expertise in this area to ensure prompt diagnosis and access to adjuvant therapies such as plasma exchange. ANCA assays are vital in the diagnosis, and patients with suspected lung haemorrhage should be tested within 24 hours of presentation. This will help to minimise morbidity and mortality from these diseases. Good long term survival can be achieved with prompt treatment with appropriate immunosuppressive agents. Continued low dose immunosuppression and follow up are required long term as there is a high incidence of relapse.