Article Text

Abstract
The management of severe pulmonary hypertension associated with right ventricular failure is reviewed and its relevance to adults with acute respiratory distress syndrome (ARDS) is discussed.
- critical care
- right ventricular failure
- pulmonary hypertension
- pulmonary embolism
- acute respiratory distress syndrome
Statistics from Altmetric.com
- critical care
- right ventricular failure
- pulmonary hypertension
- pulmonary embolism
- acute respiratory distress syndrome
The lungs are the only organs that receive the entire cardiac output which is delivered at a mean resting pulmonary arterial pressure of 15 mm Hg. The capacitance pulmonary arteries are larger in calibre and have thinner walls than their systemic counterparts. Moreover, the pulmonary circulation possesses little resting vascular tone and has a large reserve for recruitment of vascular segments that are normally non-perfused.1 Thus, the pulmonary circulation is a low pressure, low resistance circuit capable of handling large increases in pulmonary blood flow (up to sixfold with strenuous exercise) with only small changes in pressure. The maintenance of a low pulmonary capillary pressure is vital in preserving the function of the blood-gas barrier.2 In accordance with this low pressure circuit, the right ventricle (RV) is a thin muscle with limited contractile reserve, which has significant implications for both the prognosis associated with severe pulmonary hypertension (PHT) and for the principles underlying the clinical management of PHT and RV failure.
Both PHT and RV dysfunction are common complications of the complex medical disorders experienced in the intensive care unit. In most circumstances the PHT is mild or moderate in degree and associated with RV dysfunction rather than frank right heart failure. Occasionally, however, patients do present with life threatening PHT and associated RV failure requiring prompt and appropriate intervention.
Right heart dysfunction and PHT of varying severity are commonly encountered in patients with chronic lung disease and left ventricular failure, but these specific entities will not be considered further. This review will rather concentrate on the management of severe PHT in the setting of RV failure. In addition, we will discuss the relevance and treatment of PHT in the context of acute respiratory distress syndrome (ARDS) in adults. Definitions of PHT and calculations for mean pulmonary artery pressure (PAP) are shown in table 1.
Definitions of pulmonary hypertension (PHT) and calculations for mean pulmonary artery pressure (PAP)
In the intensive care unit haemodynamics are usually measured using a flow directed, balloon tipped pulmonary artery catheter. Cardiac output is most commonly and conveniently determined by thermodilution techniques. It can also be derived via the Fick principle but this is not used frequently in clinical practice. Transoesophageal echocardiography can also be used to estimate cardiac output using Doppler imaging. The procedure requires sedation, however, and is not therefore usually performed in cases with severe PHT unless the patient is intubated (or undergoing another essential procedure).
AETIOLOGY OF PULMONARY HYPERTENSION
The WHO consensus conference in 19983 reclassified PHT according to clinicopathological criteria. Both the site of pathology (arterial or venous) and causative factors (association with respiratory disease/hypoxia, thromboembolic disease, or primary vascular pathology) were included. This classification adds to our understanding of the mechanisms involved and provides a good starting point in developing a rational clinical approach to the management of severe PHT.
By this classification, the cause of PHT is divided into either an intrinsic disease of the pulmonary vessels or a vascular response to another disease process. In general, treatment for this second group should be directed at the underlying disease rather than at the pulmonary vasculature per se. Probably the commonest causes of PHT in the ITU are raised left atrial pressure and hypoxaemia. Treatment should be directed at the underlying cardiac or respiratory disease, respectively.
THE PULMONARY CIRCULATION IN ARDS
The acute respiratory distress syndrome (ARDS) is characterised by non-hydrostatic pulmonary oedema and refractory hypoxaemia, and complicates up to 25% of cases of the systemic inflammatory response syndrome The consensus definitions of ARDS and acute lung injury (ALI) are shown in table 2. PHT with increased pulmonary vascular resistance is common, even when systemic vascular resistance is low. The degree of pulmonary arterial hypertension is usually mild to moderate but promotes the accumulation of extravascular lung water and can cause right ventricular dysfunction, reducing ejection fraction and cardiac output. The presence of PHT has been shown to be an adverse prognostic indicator in patients with ARDS.4,5
Recommended criteria for definition of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS). Modified from Bernard et al42
Initially, a number of factors may contribute to the increase in PAP in ARDS.6 Increased circulating levels of vasoactive mediators such as serotonin, endothelin-1, thromboxane and leukotrienes may contribute to the increase in pulmonary vascular tone. There may also be an important contribution from increased discharge from the sympathetic nervous system. Although hypoxic pulmonary vasoconstriction may play some role in increasing pulmonary vascular resistance locally, generally the pulmonary vascular response to hypoxia is reduced in patients with ARDS.7 Indeed, administration of 100% oxygen to patients with ALI does not significantly alter pulmonary haemodynamics.8 Structural changes in small pulmonary arteries (pulmonary vascular remodelling) develop in patients with ARDS of more than a few days duration, with the severity of the changes correlating with the duration of lung injury. Initially, acute endothelial injury and thromboemboli are visible on histopathological examination.9 Fibrocellular intimal obliteration of arteries, veins, and lymphatic vessels occurs in those surviving more than 10 days.
Manipulation of the pulmonary circulation in patients with ARDS is aimed at improving systemic oxygen availability and improving right ventricular dysfunction.10 Reducing pulmonary vascular resistance using vasodilators such as prostacyclin11 or nitrates improves cardiac output in ARDS, but concurrent vasodilation occurring in poorly ventilated lung regions can worsen ventilation/perfusion (V/Q) matching, increasing shunt fraction and exacerbating hypoxaemia. The aim of inhaled vasodilator treatment is to confine vasodilation to those areas of the pulmonary circulation receiving the most ventilation, optimising V/Q matching. Thus, inhaled nitric oxide (NO) reduces pulmonary vascular resistance, improves right ventricular function, and improves arterial oxygenation in patients with ARDS.12,13 The current recommendation is that NO treatment in ARDS should be limited to patients who are optimally ventilated and have an arterial oxygen tension (Pao2) of <12 kPa with a fractional inspired oxygen (Fio2) of 1.0.14 Similar improvements have been reported with the use of nebulised prostacyclin in ARDS. However, large randomised trials have not shown that selective pulmonary vasodilators alter the outcome in ARDS in terms of a reduction in either the duration of mechanical ventilation or mortality.15
SEVERE PULMONARY HYPERTENSION
In general, the clinical presentation of severe PHT reflects the degree of resulting right heart dysfunction or failure. The structure and geometry of the RV dictates its ability to cope with increased PAP.
If the pressure increase occurs over a long period of time (months to years), the RV has a limited capacity to cope via hypertrophy. By contrast, after severe acute rises in pulmonary vascular resistance—for example, following a massive central pulmonary embolism—the ability of the RV to adapt is severely limited. Usually it simply dilates and can rarely generate, nor less sustain, a systolic pressure of 50 mm Hg.16 This results in a low output and haemodynamic shock. Occasionally in this acute setting the venous pulses, especially the femoral pulse, will become “arterialised” due to marked tricuspid regurgitation and, with the accompanying systemic hypotension, can be mistaken for its arterial counterpart.
Right ventricular failure is also manifest as systemic venous hypertension. This results in the classical clinical signs of jugular venous engorgement (with “cV” waves due to tricuspid regurgitation), pulsatile hepatomegaly, lower limb oedema, and occasionally anasarca. In addition to these clinically obvious effects there is also a concomitant congestion of the splanchnic circulation in general, with resulting gut oedema. In these circumstances patients often become refractory to oral diuretics, presumably due to poor absorption.
Other less usual presentations may develop in patients with severe primary pulmonary hypertension (PPH) attributable to the effects of venous hypertension. Renal dysfunction is common, resulting from both arterial hypotension and the high renal venous pressure. This reduces the filtration pressure across the glomerulus and may result in nephrotic syndrome as the presenting clinical manifestation of PHT. Similarly, visual disturbances associated with papilloedema and delirium due to severe cerebral venous hypertension mimicking cavernous sinus thrombosis may occur. In all cases effective treatment of the PHT leading to improvement in RV function and reduction in systemic venous pressures can lead to resolution.
Investigation of severe PHT
In most cases severe PHT can be readily diagnosed on clinical grounds. Where right heart failure dominates the clinical picture, severe PHT is usually evident. However, it is imperative to elucidate the underlying cause in order to administer appropriate treatment.
Patients with severe PHT and RV failure are frequently too unwell or too unstable to undergo definitive investigations. Where feasible, spiral CT angiography is the investigation of choice for the diagnosis of acute central pulmonary embolism. This examination will identify large central clots with positive and negative predictive values of over 90% compared with pulmonary angiography.17 Radiological signs of severe PHT such as enlargement of the pulmonary arteries and right sided chambers are also evident. Conversely, the left sided chambers are normal or reduced (compressed) in size.
Echocardiography at the bedside may show signs of RV pressure overload as well as excluding other causes of haemodynamic collapse such as pericardial tamponade.18 With severe RV dilation and dysfunction, left ventricular function as assessed by echocardiography is usually reduced due to paradoxical movement of the septum and distortion of normal left/right heart dynamics. It is vitally important in this setting to attribute correctly the low cardiac output state to the right sided problems as there are important differences in the principles of treating a failing RV and those used to support the left side.
Pulmonary artery catheterisation will confirm a low cardiac index in association with a raised pulmonary vascular resistance. A low pulmonary artery capillary occlusion pressure (<15 mm Hg), reflecting reduced left atrial pressure, effectively excludes left heart dysfunction as the cause of PHT and the low output state. However, in patients with severely deranged haemodynamics, accurate placement of a pulmonary artery catheter can be difficult and the data obtained need to be interpreted with caution.
TREATMENT
General principles
Pulmonary hypertension (PHT)
The principles of treating PHT are based upon reducing RV afterload and preventing or treating the complications of RV dysfunction. Attempted correction of hypoxaemia is mandatory in the treatment of any patient with severe PHT. In the presence of a right to left shunt this may not be possible, but in these situations it is usually possible at least to ameliorate the added oxygen desaturation associated with sleep and exercise. As in any low cardiac output state, anticoagulation is desirable.
With intrinsic disease of the pulmonary arteries, several vasodilator strategies can be considered. For patients requiring mechanical ventilation inhaled NO can be used to maximise V/Q matching, as vasodilation only occurs in ventilated areas.19 Prostacyclin (PGI2) is also an effective pulmonary vasodilator but, when administered intravenously, can result in worsening of V/Q matching.20 The short biological half life of inhaled NO (seconds) leads to a greater effect on the pulmonary than the systemic circulation, its biological effects being rapidly eliminated by reaction with haemoglobin. Prostacyclin has a rather longer half life in the circulation (1–2 minutes) and usually lowers pulmonary and systemic vascular resistance, although the systemic effects can be minimised by inhaled treatment. Calcium channel blockers should not be used in patients with significant cardiac dysfunction as their negative inotropic effects may further impair RV performance.21 Nitric oxide donors such as nitroprusside or nitrates are rarely effective in this setting, and usually exacerbate the systemic hypotension associated with the low cardiac output syndrome.
Right heart dysfunction
The RV is designed to work with a low pressure circuit and, as such, has limited contractile reserve. For this reason RV support is aimed at reducing afterload. Severe acute RV dysfunction associated with PHT may present during heart or lung transplantation or surgery for pulmonary embolus or severe mitral valve disease. Inhaled NO may be useful in this setting to decrease pulmonary vascular resistance without reducing systemic arterial pressure, which is essential for the maintenance of coronary perfusion to the right ventricle. Inhaled NO in the dose range 20–40 ppm may benefit these patients.14 Unless the pulmonary vascular resistance can be reduced, inotropes are ineffective, imposing more work on a struggling RV. Inotropes can transiently improve cardiac output for 6–12 hours but inevitably the RV fails, resulting in a lethal downward spiral of increasing inotrope requirements but diminishing effect. Inotropes are, however, useful for supporting or augmenting RV contractility in situations where the pulmonary vascular resistance can be reduced through concomitant administration of vasodilator therapy. In this respect, a phosphodiesterase inhibitor such as enoximone22 is our preferred choice as its vasodilator properties contrast with the pulmonary vasoconstrictor effects characteristic of catecholamines.
Intra-aortic balloon counterpulsation is useful for short term RV support,23 augmenting coronary blood flow and increasing central systemic blood pressure, thereby reducing the need for pressor agents such as noradrenaline which are potent pulmonary vasoconstrictors. Most devices used in the context of RV support are modified from those used to support the failing left heart. When the lung function remains adequate to allow sufficient oxygenation and carbon dioxide removal, mechanical assist devices alone may be used to support the RV. When severe lung injury accompanies RV failure, mechanical RV support can be incorporated into either extracorporeal membrane oxygenation (ECMO) or extracorporeal carbon dioxide removal (ECCOR) circuits.
Paracorporeal devices such as the Abiomed are implanted through a sternotomy using right atrial or ventricular cannulation for drainage to the pump chamber, with return to the pulmonary artery through a vascular conduit sutured to the pulmonary trunk. Such a procedure is highly invasive and the subsequent management of the pump is complex. In particular, maintaining a balance between vasodilator agents, inotropes, and filling pressures may be difficult to achieve. Overflowing the pump can result in gross pulmonary oedema, compounding an already difficult situation. A further source of difficulty may be experienced with the management of anticoagulation. While it is important to achieve adequate anticoagulation, intracranial haemorrhage may result if tight control is not maintained. Success is most likely in centres where there is familiarity with the management of these devices and their complications.
Although the use of ECMO is more complex, cannulation and the establishment of the ECMO circuit is less invasive than that required for most paracorporeal pulsatile ventricular assist devices. This technique is only of value when the insult necessitating its use is reversible. Although well established in children, its adoption in adult practice has been less widespread. Key components to successful intervention include early institution (ventilated less than 5 days), a flexible approach to cannulation (established percutaneously via femoral and jugular routes), and readiness to support other systems including the kidneys and liver.
Any attempt to support the RV with a mechanical device in the face of PHT is complicated. A multidisciplinary approach must be adopted, and there is no simple unifying technique. The advent of small axial and rotary blood pumps currently undergoing trials in left ventricular dysfunction may improve the outlook in this area in years to come.
Atrial septostomy has been used in the treatment of patients with severe PHT and RV compromise.24 This treatment evolved from the observation that patients with PPH and a patent foramen ovale lived longer than those with no septal defect.25 Creating a shunt at the atrial level decompresses the right sided chambers and augments left atrial filling with a concomitant increase in cardiac output and systemic oxygen transport. The resulting reduction in RV end diastolic pressure and wall tension are postulated to improve Starling haemodynamics and RV contractility. Although the resulting right to left shunt causes systemic oxygen desaturation (which is especially marked with exercise when PAP rises), this can usually be controlled with supplemental oxygen.
The clinical benefits reported following septostomy include resolution of syncopal and pre-syncopal episodes, decreased cough, decreased systemic venous congestion, and improved exercise tolerance. Our own experience of this procedure suggests it is most effective before the onset of severe RV dysfunction and, conversely, as reported by others,26 it has not been effective in patients with end stage right heart failure or acute right heart failure severe enough to require admission to the ITU. Atrial septostomy has been used primarily in advanced PPH as a bridge to heart-lung transplantation,24 although its exact role (in particular the optimal timing of the procedure) is not known. As our own experience with this procedure has developed, it is being used earlier in the course of disease. Experience of the procedure in any form of severe PHT is very limited and at present there are no controlled clinical studies in any disease group.
Thromboembolic disease
The most common cause of acute severe PHT is massive central pulmonary thromboembolism. Some of the mechanisms involved in the development of shock in the setting of acute massive pulmonary embolism (PE) are shown in fig 1. The overall mortality from massive PE is 6–8%, increasing to 30% if complicated by systemic hypotension.27 Of those patients who fail to survive, 67% die within 1 hour of the onset of symptoms.28

Factors involved in the generation of acute right ventricular failure following massive pulmonary embolism. MPAP=mean pulmonary artery pressure; CO=cardiac output; LV=left ventricle; Pvo2=ventricular oxygen tension.
The diagnosis of massive PE is suggested by the clinical presentation of right ventricular failure, a normal or oligaemic chest radiograph, and a suggestive ECG (right ventricular strain).29 In more stable patients there may be time to organise spiral CT pulmonary angiography.17,30 Echocardiography (either transthoracic or transoesophageal) may reveal thrombus in the pulmonary outflow tract or show signs of right ventricular dysfunction/hypokinesis.18,31
Oxygen and analgesia should be given to all patients immediately. Invasive monitoring of the central venous pressure will guide cautious fluid and colloid replacement to optimise right sided filling pressures. The central venous pressure should be maintained at 15–20 cm H2O. Overfilling worsens right ventricular function, but inadequate filling (or indeed overly aggressive diuresis) also compromises RV haemodynamics. If haemodynamic compromise is present and there are no contraindications (shown in box 1), thrombolysis should be considered for acute massive PE.32,33 The rationale for this is the greater mortality in patients with right ventricular dysfunction following acute PE.34 Thrombolysis leads to more rapid restoration of RV function than heparin alone.28 However, the potential benefits must justify the 1% risk of cerebral and fatal bleeding,35 and the effects of thrombolysis on mortality still need to be confirmed by a prospective randomised trial. Two hour infusion regimens of streptokinase (1.5 million units), urokinase and recombinant tissue plasminogen activator (rt-PA; 100 mg) followed by a heparin infusion have similar efficacy and safety profiles.36 Thrombolysis may be considered in all age groups and in postoperative patients. The risk of major haemorrhage with these agents increases with increasing age and body mass index. Bolus and front loaded regimens (administered over <2 hours) are simpler to use and are as effective as longer duration infusions. Streptokinase should not be used if patients have been previously treated with this agent. There is no benefit of direct central versus peripheral administration of thrombolytic agents.
Box 1 Indications and contraindications for thrombolytic treatment of acute PE
Indications
-
Massive PE: first line treatment
-
Haemodynamic compromise
-
Failure to respond to anticoagulants
Contraindications
Absolute
-
Recent major trauma or operation (within 10 days)
-
Recent cerebrovascular accident (within 2 months)
-
Bleeding diathesis
-
Active internal bleeding
Relative
-
Prolonged cardiopulmonary resuscitation
-
Pregnancy
-
Diabetic proliferative retinopathy
Patients may require inotropic treatment as the RV afterload is reduced and RV function recovers.37 It is important to maintain systemic arterial pressure and to ensure adequate perfusion of the right coronary artery.
Studies have now shown that echocardiography can also provide important prognostic information in acute PE. Although cardiogenic shock occurs in less than 5% of patients with PE, RV dysfunction or hypokinesis occurs in up to 40% of patients with a normal systemic blood pressure. The finding of RV dysfunction increases the mortality from PE at 14 days and 3 months. In a Swedish study34 mortality at 1 year was 45% in patients presenting with RV dysfunction compared with 15% in those with normal echocardiography. The benefits of thrombolysis may therefore extend to patients without overt shock but with RV dysfunction of any degree. This was borne out by the results from a multicentre registry showing a favourable clinical outcome in haemodynamically stable patients with major PE following thrombolysis.38 Again, these promising results await confirmation in a prospective randomised trial.
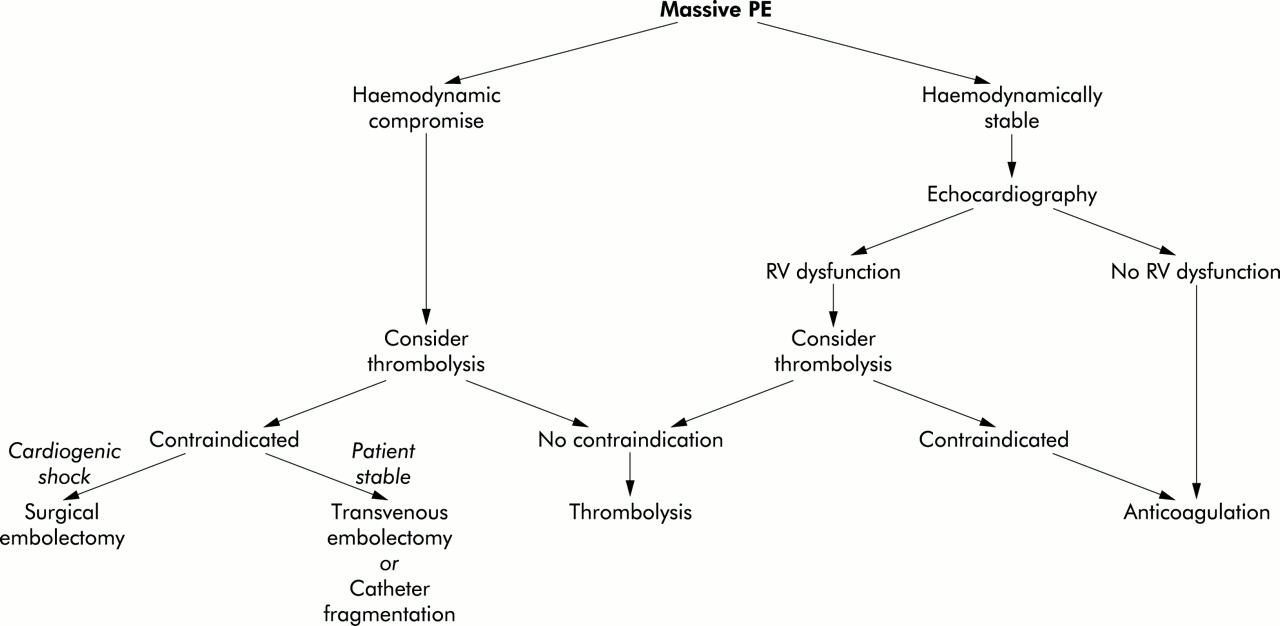
If thrombolysis is contraindicated (box 1) in acute massive PE, other treatment options include mechanical clot fragmentation, transvenous catheter embolectomy, or surgical embolectomy.33 Insertion of an inferior vena caval filter should be considered in the presence of active haemorrhage to prevent further potentially fatal embolism. Surgical embolectomy may be appropriate in the setting of an experienced cardiovascular surgical unit. It should be reserved for severely compromised patients in refractory cardiogenic shock or patients requiring intermittent resuscitation.39 To be effective it must be performed as soon as possible. Total perioperative mortality is approximately 30%, with the highest mortality rates (∼60%) in those patients who require preoperative cardiopulmonary resuscitation.40,41 An approach to the treatment of a patient with massive PE is shown in fig 2. In this setting, it is vitally important that chronic thromboembolic disease is excluded as the treatment of these two conditions is vastly different. Attempted surgical embolectomy (as opposed to pulmonary endarterectomy) in a patient with chronic thromboembolic PHT is fraught with disaster.
{kind=link}
{kind=link}
Therapeutic approach to massive PE.
For patients with chronic thromboembolic PHT, the treatment of choice is pulmonary endarterectomy. In experienced hands this procedure results in a sustained reduction in pulmonary pressures and RV remodelling.43 Within the UK, Papworth Hospital is the National Specialist Commissioning Advisory Group (NSCAG) designated centre for pulmonary endarterectomy.
CONCLUSION
Unrecognised or untreated severe PHT has a poor prognosis related directly to the limited contractile reserves of the RV. Appropriate treatment hinges on identification of the underlying cause and effective reduction in RV afterload. The commonest cause of acute severe PHT is massive pulmonary thromboembolism and, if no contraindications exist, thrombolytic therapy is the treatment of choice. Pulmonary vasodilators such as intravenous prostacyclin or inhaled NO are often effective in other cases where increased pulmonary vascular tone is present. Whatever the underlying cause, without effective afterload reduction the RV will inevitably fail and it is thus of the utmost importance that inotropes are not relied on to support the RV without effective treatment of the underlying problem.