Article Text

Abstract
Pharmacological approaches to the treatment of ARDS are reviewed. Future treatments should be targeted at elements of the pathological process that produce specific clinical problems.
- acute respiratory distress syndrome
- critical care
Statistics from Altmetric.com
Our understanding of the pathophysiology and management of the acute respiratory distress syndrome (ARDS) has improved immensely since its original description, but pharmacotherapies have proved disappointing in clinical trials. Several reasons have been proposed for this failure.
-
There are no good experimental models of ARDS so that drugs may not have the desired effect or produce unacceptable side effects when used clinically.
-
The inflammatory cascades that cause sepsis and ARDS are characterised by widespread redundancy so it is unlikely that a single agent could reverse or terminate such complex processes.
-
Although a drug may improve pulmonary function, it may not alter outcome. Fewer than 5% of patients with ARDS die of respiratory failure; the majority suffer from multiple organ failure and succumb after withdrawal of support.
-
Enrolling patients with ARDS in clinical trials using the American-European Consensus Conference definition1 ignores the heterogeneity of the disease. In pathological terms, the acute exudative and fibroproliferative phases present distinct targets for intervention, making the timing of drug administration (after disease onset) crucial. The primary cause(s) of ARDS, the patient's age and medical history all affect prognosis and possibly drug responsiveness of the condition.
Recent reports suggest that the mortality associated with ARDS may be falling, probably because of advances in supporting critically ill patients.2 This trend may increase the number of potential survivors and the window of opportunity for pharmacological manipulation of lung injury. Thus, drugs that have been apparent failures in terms of mortality may still have a useful role to play, either in combination with other agents or in subgroups of ARDS patients defined either by their underlying condition or by their stage of lung injury. This article reviews the major pharmacological approaches to treating ARDS in the context of modern supportive care (fig 1). We conclude with a proposal for future strategies in the non-ventilatory management of ARDS.
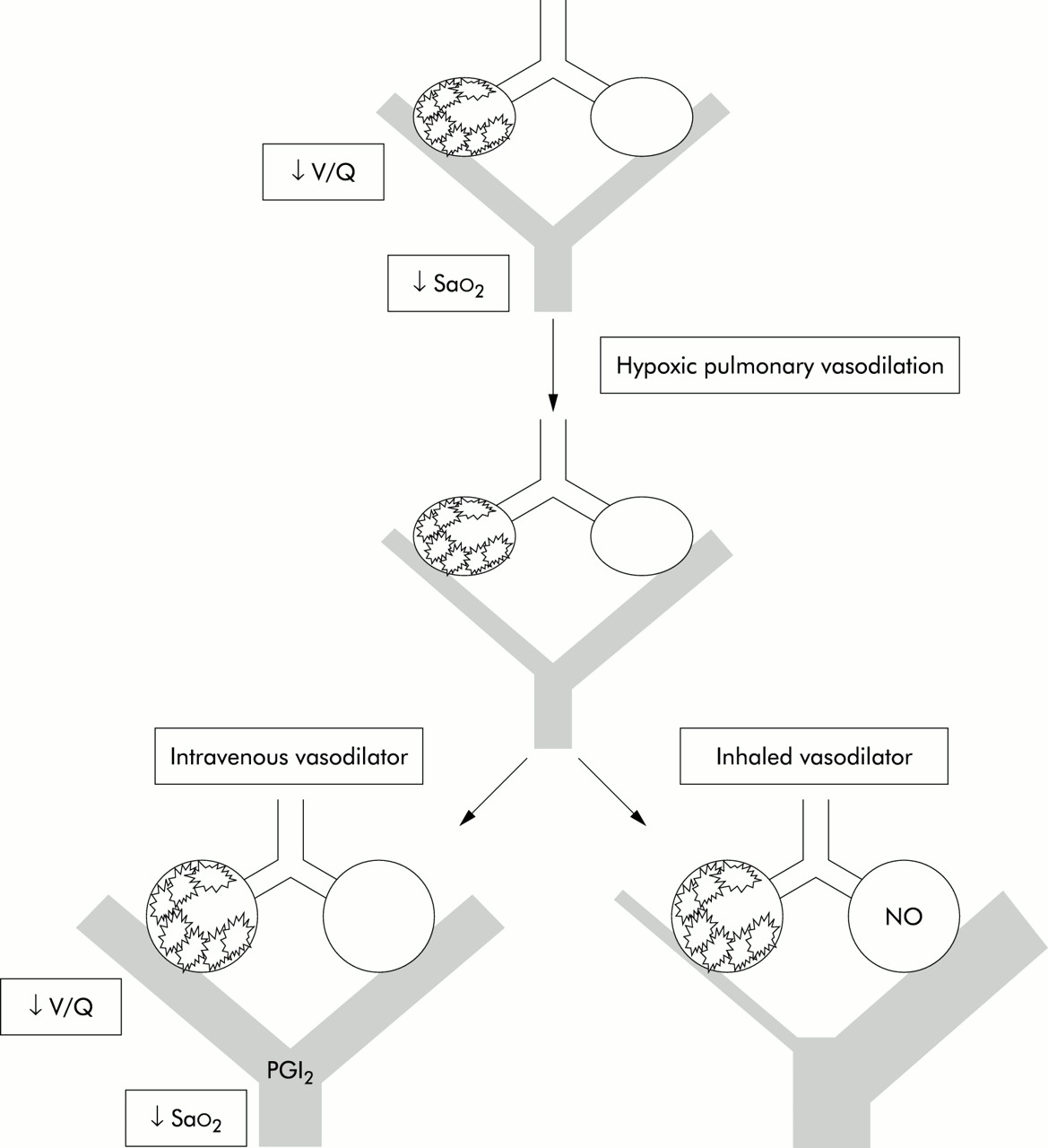
Effect of intravenous and inhaled vasodilators in lung injury. V/Q=ventilation:perfusion ratio; Sao2=arterial oxygen saturation; PGI=prostacyclin; NO=nitric oxide.
INTRAVENOUS FLUID MANAGEMENT
Patients with ARDS often have cardiovascular dysfunction caused by systemic inflammation that is commonly associated with sepsis. Hence, myocardial depression, abnormal vascular tone, and permeability contribute to abnormal tissue oxygenation and ultimately organ failure. In practice, achieving adequate organ perfusion may occur at the cost of increasing extravascular water manifesting as an exacerbation of pulmonary oedema. Low extravascular lung water levels are associated with better oxygenation and a lower mortality in patients with ARDS in retrospective studies.3,4 There is limited prospective evidence that targeting lower extravascular lung water using diuretics with vasopressors to support organ perfusion reduces the time required on a ventilator.5,6 Our policy is to keep the intravascular volume as low as possible while maintaining an adequate cardiac index and mean arterial pressure.
INHALED VASODILATORS
Nitric oxide (NO) is a free radical gas produced constitutively in the lung by nitric oxide synthase from l-arginine, NADPH, and oxygen. Endothelial cells constitutively release NO, causing pulmonary vasodilation primarily via the secondary messenger cyclic guanosine monophosphate. ARDS is characterised by ventilation-perfusion mismatching which produces arterial hypoxaemia that may in part be caused by disordered endogenous NO activity. Patients with ARDS commonly have mild pulmonary hypertension. Any inhaled vasodilator can augment hypoxic pulmonary vasoconstriction by selectively vasodilating vessels associated with ventilated alveoli to improve oxygenation (fig 2).

{kind=link}
{kind=link}
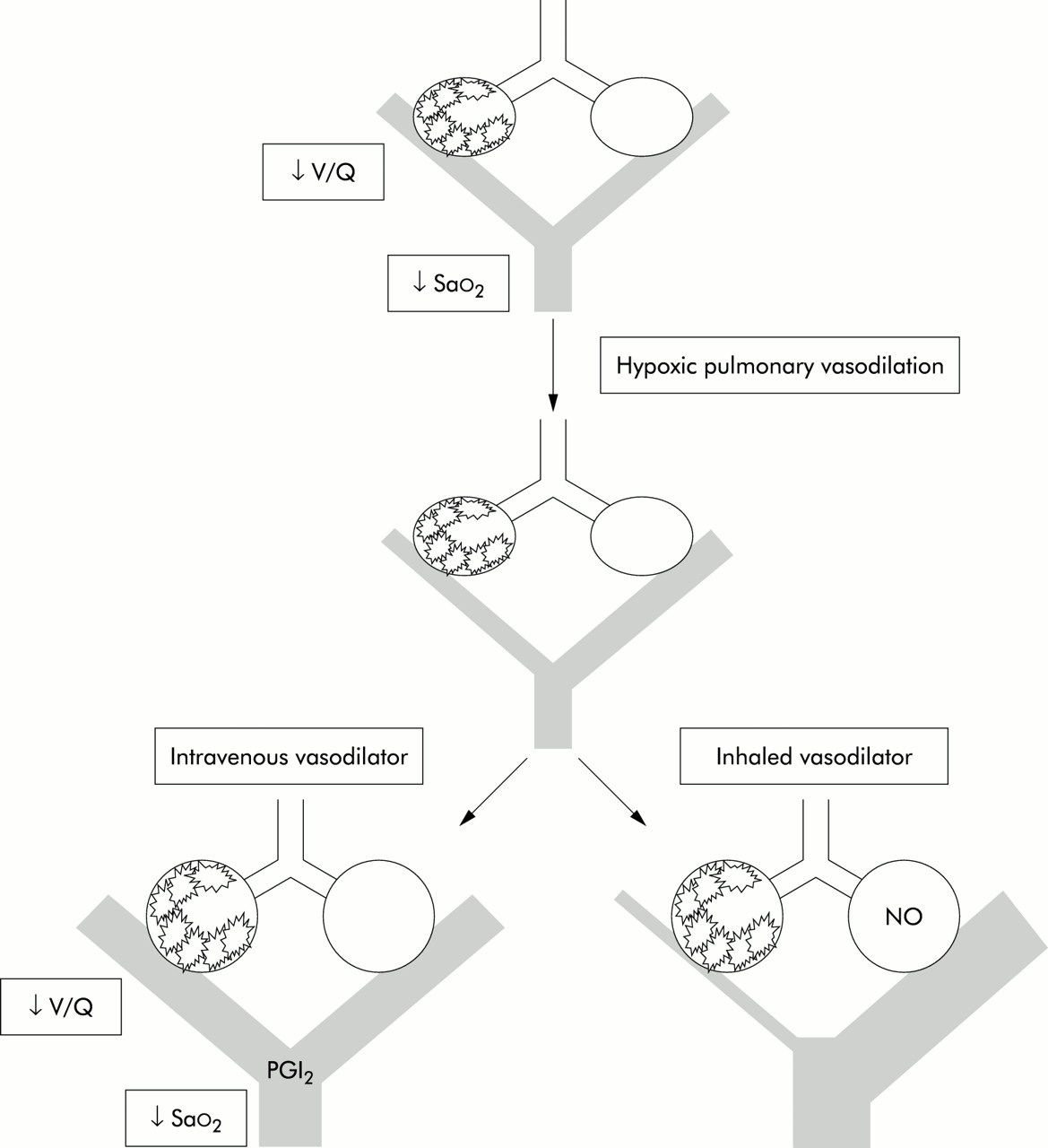
Suggested treatment algorithm for acute lung injury (ALI) and ARDS. AECC=American & European Consensus Conference 1993; PAC=pulmonary artery catheter; CT=computed tomography; BAL=bronchoalveolar lavage; Fio2=fractional inspired oxygen concentration; Sao2=arterial oxygen saturation.
Improved oxygenation and direct vascular smooth muscle relaxation by NO also reduce pulmonary vascular resistance (PVR). Vasoconstriction by hypoxia, hypercapnia, thromboxane A2, and angiotensin II can all be partially reversed by inhaled NO, although the PVR of normal volunteers is not affected.7 Reducing PVR and consequential improvements in right ventricular function may benefit some patients with ARDS. However, NO does not increase cardiac output in the majority.8 Reduced arteriolar and venous tone may lower capillary pressure, reducing leakage and further improving gas exchange. Unfortunately, in patients with pulmonary hypertension associated with impaired left ventricular function, pulmonary vascular relaxation may also increase pulmonary oedema.
Endothelial NO also inhibits platelet aggregation and neutrophil adhesion that are possible mediators of lung injury. Although the importance of these actions in ARDS is uncertain, inhaled NO has been used immediately after lung transplantation to reduce ischaemia-reperfusion injury.9 NO is generally unreactive. However, it quickly reacts with other radicals and reactive oxygen species (ROS) including the superoxide anion to produce more reactive products such as peroxynitrite. Although ROS are usually kept at low levels in lung tissue by antioxidants and dismutases, these protective systems may be overwhelmed during ARDS.10 In vitro, peroxynitrite oxidises and nitrosylates proteins, nucleic acids and lipids, including essential components of the surfactant system. However, the clinical significance of peroxynitrite production is unknown.
Approximately 60% of patients with ARDS or acute lung injury (ALI) of all causes respond to inhaled NO, increasing their Pao2 by more than 20%.11 The effect can frequently be seen in less than 10 minutes or may take several hours.12,13 However, in several trials the oxygenation of control groups has risen to meet that of NO treated patients between 24 hours and 4 days.11,13,14 The concentration-response relationship between inhaled NO and arterial oxygenation shows considerable interindividual variation.15 Currently there are no indicators that will predict the response. Maximal improvement in oxygenation is sometimes achieved with 1–2 parts per million (ppm) and occurs at less than 10 ppm in most patients. Maximal reduction in pulmonary artery pressure is usually obtained between 10 and 40 ppm, with no benefit and possible toxicity at doses greater than 80 ppm. United Kingdom guidelines suggest a maximum dose of 40 ppm.16 Intra-individual variation in response with time is also significant and may be influenced by lung recruitment, co-existent pathology, or the resolution of inflammation. Clinically, it is sometimes difficult to stop inhaled NO without “rebound” pulmonary hypertension and hypoxaemia. The last 1–2 ppm may have to be weaned especially slowly.
The systemic effects of inhaled NO are negligible due to the rapid strong combination of NO with haemoglobin to form methaemoglobin. This is normally reduced to functional haemoglobin and NO is ultimately converted to soluble NO3. Methaemoglobinaemia produces a functional anaemia and a left shift in the dissociation curve, but rarely causes a clinical problem. Normal levels of methaemaglobin are less than 2% and values less than 5% usually do not need treatment. NO reacts slowly with oxygen and water to form toxic NO2, nitrous and nitric acids. These damage the lung at concentrations as low as 2 ppm. The reaction rate is proportional to the fractional inspired oxygen concentration (Fio2) and the square of the NO concentration. Thus, the contact time and concentrations of the gases should be kept to a minimum. With proper monitoring, delivery systems, and NO doses of less than 40 ppm, NO2 is not a significant problem. Delivery systems that add NO “upstream” of the ventilator allow longer mixing with oxygen and are not recommended. Continuous “downstream” addition of NO may allow NO to collect in the inspiratory limb of the circuit during the expiratory phase of some systems. Synchronised NO delivery during inspiration may be the optimum mode of delivery. NO contained in exhaled gas should be absorbed before release.
Randomised controlled trials in patients with ARDS have shown that, while inhaled NO temporarily improves oxygenation and reduces pulmonary artery pressure in the majority, its use is not associated with an improved outcome (table 1). Inhaled NO is therefore not a standard treatment for ARDS. However, patients with severe refractory hypoxaemia and inadequate right ventricular function secondary to pulmonary hypertension may benefit from inhaled NO. NO may also protect patients whose oxygenation might otherwise depend upon a potentially damaging ventilatory strategy. In small studies the pulmonary vasoconstrictor almitrine improved oxygenation in patients with ARDS.17 It has been suggested that low dose intravenous almitrine potentiates hypoxic pulmonary vasoconstriction and the combination of almitrine and inhaled NO may improve oxygenation synergistically in patients with ARDS.18
Randomised controlled trials of inhaled nitric oxide (NO) in patients with ARDS
Prostacyclin (PGI2) is an endothelium-derived prostaglandin vasodilator that inhibits platelet aggregation and neutrophil adhesion. Its mechanism of action differs from NO in that smooth muscle relaxation is associated with a rise in cytoplasmic cyclic adenosine monophosphate. Its half life is only 2–3 minutes but it is not metabolised by the lung, so when administered intravenously, PGI2 lowers pulmonary vascular resistance but may also increase intrapulmonary shunting and cause systemic hypotension.15 However, nebulised PGI2 (0–50 ng/kg/min)19,20 or alprostadil (PGE1, 20–80 μg/h)21 produce equivalent effects to inhaled NO with minimal systemic side effects and without measurable platelet dysfunction, but there have been no large randomised trials of these therapies. Nevertheless, the relatively simple delivery system, harmless metabolites, and no requirement for special monitoring make nebulised PGI2 an attractive alternative to inhaled NO, despite its expense.
CORTICOSTEROIDS
Corticosteroids reduce the production of a great number of inflammatory and profibrotic mediators by many mechanisms. The importance of steroid therapy to the resolution of lung inflammation in animal models became apparent in the 1980s. Unfortunately, trials of short term, high dose steroid therapy (for example, methylprednisolone 30 mg/kg 6 hourly for 24 hours) failed to show an improvement in mortality of patients at risk of or with early ARDS associated with sepsis, aspiration, and trauma (table 2).22–26 In fact, some trials showed increased risk of infection, lower rates of reversal of ARDS, and increased mortality associated with the use of high dose steroids. Meta-analyses of available trials emphasise the adverse effects of these agents in patients with sepsis.
Published trials of short term, high dose steroid therapy in patients with ARDS
However, the use of steroids in patients with late ARDS (7–14 days from diagnosis) has not been abandoned. Recent data suggest that inflammation and fibrosis in the lung are distinct and thus independently manipulable processes.27 There is also clinical evidence that steroids favourably modify the fibroproliferative phase of ARDS. In the 1990s lower dose (2–8 mg/kg/day methyprednisolone), longer term (2–6 weeks) corticosteroid treatment was used in patients with ARDS of over 10 days duration. Mortality fell to approximately 20% in some uncontrolled series but complications attributable to steroids were not infrequent and included sepsis, pneumonia, wound infection, gastric ulceration, and diabetes. A trial using this approach randomised 24 patients with “unresolving” ARDS of more than 7 days duration to methylprednisolone (2 mg/kg load then 2 mg/kg/day in four divided doses reducing weekly to 1 mg/kg/day, then 0.5 mg/kg/day, then 0.15 mg/kg/day).28 None of the 16 patients in the steroid group died compared with five of eight originally given placebo. Steroid therapy was associated with improved oxygenation and successful extubation. Eligible patients were examined for pulmonary infection by bronchoalveolar lavage at day 5 and all febrile patients received a broad septic screen before trial entry. Documented infections were treated with appropriate antibiotics for at least 3 days before steroids were administered. Despite these precautions, 75% of patients in both arms of the trial suffered new sepsis. Although the results are promising, the design and interpretation of this trial have proved contentious.29 The larger NIH ARDS Network Late Steroid Rescue Study may provide sounder evidence for prescribing low dose steroids in late ARDS.
SURFACTANT
Type II alveolar cells synthesise and recycle surfactant phospholipids and proteins. Surfactant lowers alveolar surface tension and prevents collapse at low lung volumes. The same effects reduce the hydrostatic pressure gradient favouring fluid movement into the alveolar space. Surfactant also has anti-inflammatory and antimicrobial properties. During ARDS, surfactant activity may be deficient because of reduced production, increased removal with recurrent alveolar collapse during ventilation, abnormal composition and, importantly, dysfunction caused by plasma proteins,30 ROS, and proteases in the flooded alveolar space.31
Various preparations, doses, administration regimens, and delivery techniques have been proposed. Phospholipids alone are inferior to composites of lipid and surfactant proteins. The amount of drug required varies with the type of administration technique—intratracheal delivery, aerosolisation in ventilator gas, and direct bronchoscopic instillation. Simultaneous segmental lavage has been combined with the latter approach and this process itself may benefit some patients. The optimum timing and duration of surfactant therapy is still to be determined.
A phase III clinical trial of one aerosolised preparation in sepsis induced ARDS has been conducted on a background of improved oxygenation in previous reports.32 No significant effect was seen on oxygenation, duration of ventilation, or survival with up to 5 days of continuous treatment beginning within 48 hours of ventilation. However, this study has been criticised because the compound used contained no protein and aerosolisation may have delivered less than 5 mg/kg/day of phospholipid when investigations suggest instillation of 300 mg/kg/day may be required. Bovine surfactant administered intratracheally in a smaller trial has shown that higher doses (100 mg/kg qds) are required to alter alveolar surfactant composition.33 This dose improved oxygenation over 120 hours and a trend was observed towards reduced mortality at 28 days. Total bronchopulmonary segmental lavage with 30 ml per segment of a synthetic protein containing surfactant was safe and effective at improving oxygenation within 72 hours of the onset of sepsis induced ARDS.34 Despite these trials, the use of surfactant in ARDS remains experimental. Successful use of surfactant in animal models of lung injury and in neonatal respiratory distress syndrome suggests that efficient administration of an effective substitute could be beneficial in ARDS. The goals of treatment would include both improved gas exchange and protection against ventilator induced lung injury.
IMMUNONUTRITION
Avoiding nutritional depletion while delivering a high fat, low carbohydrate diet to reduce carbon dioxide production and ventilatory demand is appropriate for patients with ARDS.35 Immunonutrition aims to influence inflammation positively and to protect gastrointestinal integrity. However, supplying enteral nutrition of any type may stimulate gastrointestinal and pulmonary IgA defence mechanisms.36 In vitro, dietary additives improve depressed immune cell function from some critically ill patients and attenuate the production of proinflammatory mediators in others.
The amino acids glutamine and arginine may be useful dietary additives for patients at risk of or with established ARDS.37 Enterocytes metabolise glutamine in a manner that enhances intestinal mucosal integrity and reduces translocation of bacteria and toxins into the portal circulation that may fuel a systemic inflammatory response. Glutamine and arginine also augment lymphocyte function, and arginine improves monocyte function in critically injured patients. l-arginine supplementation may also increase NO production, alter vascular tone, and augment free radical mediated antibacterial defences. Similarly, omega-3 fatty acids such as eicosapentenoic acid and the unsaturated oil gamma-linolenic acid reduce proinflammatory cytokine and eicosanoid production.38 Less biologically active eicosanoids such as prostaglandin PGE1, thromboxane TXA3, and leukotriene LTB5 are produced from these unsaturated fats by cyclo-oxygenase (COX) and 5-lipoxygenase during inflammation. Animal experiments suggest that polyunsaturated fatty acids can reduce pulmonary vascular resistance, lung neutrophil infiltration, and microvascular permeability, thereby improving gas exchange.
In patients with ARDS, enteral immunonutrition supplemented with antioxidants for at least 4 days was associated with reduced pulmonary neutrophil recruitment, improved oxygenation, a shortened duration of mechanical ventilation, and reduced morbidity in terms of new organ failure.39 However, there was no difference in mortality between the control and treatment groups. A meta-analysis of 12 randomised controlled trials comparing critically ill medical, surgical, and trauma patients given standard enteral nutrition with patients receiving immunonutrition suggested reduced rates of infection including nosocomial pneumonia, but again no effect on mortality.40 With some reservations, the authors concluded that their data suggested real benefits of immunonutrition in surgical and trauma patients, but that a large double blind, multicentre, randomised controlled trial was still required.
PROSTAGLANDIN E1
Intravenous PGE1 causes both pulmonary and systemic vasodilation and, in some critically ill patients, increases cardiac output and oxygen delivery.41 Although the effect on the pulmonary circulation is usually small, vasodilation is more marked under hypoxic conditions, and the nebulised drug improves ventilation-perfusion matching.21 PGE1 also inhibits platelet aggregation and neutrophil adhesion. The initial trial of PGE142 showed improved survival in trauma patients with respiratory failure. However, this benefit could not be reproduced in a subsequent multicentre trial43 in patients suffering from lung injury precipitated by surgery, trauma, or sepsis. The dose of PGE1 was limited by side effects, particularly systemic hypotension. More recent trials have used liposome technology to increase drug delivery while mitigating side effects.44,45 The use of a liposome itself is associated with immune modulating effects including downregulation of neutrophil adhesion molecules. A combined PGE1-liposome preparation in a rodent model of ALI reduced pulmonary neutrophil infiltration and capillary leak.46 However, although the phase II and III trials of liposomal PGE1 showed that patients with ARDS receiving the drug had more rapid improvements in the Pao2/Fio2 ratio, neither a survival benefit nor a reduced requirement for ventilatory support was found in the treatment group.44,45 Retrospective subgroup analysis suggested that high dose therapy might reduce the time to extubation.
THROMBOXANE SYNTHASE AND 5-LIPOXYGENASE INHIBITORS
Thromboxane and leukotrienes are in part responsible for the pulmonary hypertension and hypoxaemia of ARDS. Pulmonary vascular smooth muscle cells, endothelial cells, platelets, and neutrophils all release TXA2 on stimulation. TXA2 can initiate microvascular thromboses consisting of neutrophil and platelet aggregates that are responsible for perfusion abnormalities and recurrent ischaemia-reperfusion injury to the lung. The vasoconstrictive effect of TXA2 similarly contributes to impaired gas exchange.47 In animal models of lung injury thromboxane synthase inhibition reduced pulmonary oedema formation and inhibited microembolism, but pulmonary hypertension was only partially relieved.48
Leukotrienes (LT) are derived from arachidonic acid by 5-lipoxygenase. LTB4 is a potent neutrophil chemokine while LTC4 and LTD4 cause pulmonary vasoconstriction, capillary leak, and pulmonary oedema. The role of leukotrienes in ARDS has been less well researched but bronchoalveolar lavage fluid from patients with ARDS contains increased concentrations of LTB4, LTC4 and LTD4, which may be markers for developing ARDS.49 Ketoconazole is an imidazole antifungal agent that inhibits thromboxane synthase and 5-lipoxygenase without inhibiting COX. Ketoconazole may therefore have a dual anti-inflammatory action in ARDS by inhibiting inflammatory eicosanoid synthesis and directing COX products down other less inflammatory metabolic paths such as those synthesising prostacyclin or PGE2.50 Four trials have used enteral ketoconazole in patients at risk of or with ARDS. The incidence of acute respiratory failure was reduced in high risk surgical patients and other critically ill patients.51–53 However, an ARDS Network trial (http://hedwig.mgh.harvard.edu/ardsnet) in patients with established ARDS of medical and surgical aetiology found no differences in in-hospital mortality, ventilator free days at day 28, organ failure-free days, or markers of gas exchange between patients given ketoconazole or placebo.54 This trial achieved plasma levels of ketoconazole higher than targeted previously but could not demonstrate a reduction in thromboxane production in vivo. The effect of decreasing thromboxane synthesis in ARDS is therefore still unknown. It is still possible that ketoconazole in surgical and trauma patients at risk of or with incipient pulmonary injury may be beneficial.
ANTIOXIDANTS
The damage caused by ROS to matrix and cellular proteins, lipids and nucleic acids and ROS mediated signalling contribute to the pathogenesis of ARDS.55 The thiol groups of glutathione concentrated in the lower respiratory tract normally provide physiological antioxidant protection. However, the concentration and activity of glutathione in bronchoalveolar lavage fluid of patients with ARDS is reduced.56 Intravenous administration does not reliably raise glutathione levels, but glutathione synthesis is stimulated by N-acetylcysteine (NAC) and procysteine (L-2-oxothiazolidine-4-carboxylate). Administration of these precursors increases plasma, erythrocyte, neutrophil, and BAL fluid concentrations of glutathione in patients with ARDS, although a complete effect may require 10 days of treatment.57 Early results of NAC therapy were promising, but several trials have found no difference in mortality, length of ventilatory support, or improvement in oxygenation in patients with established ARDS,58–61 and a large phase III trial of procysteine was stopped early because of concerns about mortality in the treatment arm of the study. Currently, there is little evidence that intravenous NAC or procysteine are of benefit to patients with ARDS.
Dietary antioxidants such as ascorbic acid, tocopherol, and flavonoids have ROS scavenging ability and the capacity to reduce oxidised antioxidants as well as binding ROS producing catalysts such as free iron. Although reports of a significant action of these supplements on pulmonary inflammation are lacking, they are already components of some “immunonutrition” preparations.39
PHOSPHATIDIC ACID INHIBITION
Phosphatidic acids are liberated in response to inflammatory stimuli by lysophosphatidic acyl transferase and, like arachidonic acid, are a source of inflammatory mediators. Pentoxifylline and its more potent metabolite lisofylline are lysophosphatidic acyl transferase inhibitors. These compounds reduce serum free fatty acids in humans and lower cytokine production, neutrophil activation, pulmonary neutrophil sequestration, and attenuate lung injury in animal models. However, a study of lisofylline in 235 patients with ALI and ARDS was stopped at the first interim analysis because the pre-specified level of improvement for the treatment arm of the trial was not achieved.62
CONCLUSIONS
Most pharmacological strategies used in ARDS have targeted the inflammatory response. Many agents not mentioned here—cytokine inhibitors, anti-inflammatory cytokines, anti-proteases, and anti-endotoxin agents—alone and in combination are in the early phases of drug development. Between 18% and 41% of patients with sepsis will develop ARDS and post mortem examination reveals evidence of infection, particularly pneumonia, in almost all patients with ARDS.63 This overlap between sepsis and ARDS means that improvements in the treatment of sepsis may influence the incidence and outcome of ARDS. Large clinical trials of anti-inflammatory agents for sepsis such as ibuprofen,64 anti-endotoxin,65,66 and anti-tumour necrosis factor alpha antibodies67 have had no impact on the mortality associated with ARDS so far. Recombinant activated protein C has been shown to decrease the mortality of patients with sepsis and may improve the outlook of patients with sepsis induced ARDS.68
If the future of ARDS treatment lies in improvements in the management of multiorgan failure, then the pharmacological approach to treating lung injury may change. Previous attempts at simply dampening or halting the whole acute inflammatory process may be replaced by targeted therapies directed at elements of the pathological process that produce specific clinical problems. For example, reducing pathological fibrosis may be possible when more is understood about the regulation of collagen turnover in the normal and injured lung. Similarly, the protection, stimulation or suppression of alveolar cell division, migration, and secretion may be possible. This may enhance the repair of the alveolar epithelial cell barrier, the clearance of intra-alveolar exudate, and the normal turnover and function of surfactant.