Article Text

Abstract
Background: Adenosine induced bronchoconstriction in patients with asthma is thought to be mediated by the synthesis and release of autacoids from airway mast cells. In vitro, adenosine induced constriction of asthmatic bronchi is blocked by a combination of specific histamine and cysteinyl leukotriene receptor antagonists, but the relative contribution of these mediators in vivo is unclear. We hypothesised that adenosine induced bronchoconstriction in asthmatic patients may be blocked by pretreatment with the orally active selective cysteinyl leukotriene-1 (CysLT1) receptor antagonist, montelukast.
Methods: In a randomised, double blind, crossover study, oral montelukast (10 mg) or placebo was administered once daily on two consecutive days to 18 patients with mild to moderate persistent atopic asthma. Incremental doses of adenosine 5`-monophosphate (AMP) from 0.39 to 400 mg/ml were inhaled by dosimeter and the dose producing a 20% fall in FEV1 (PC20AMP) after AMP inhalation was recorded. Leukotriene E4 (LTE4) urinary concentrations were measured by enzyme immunoassay 4 hours after AMP challenge.
Results: Montelukast pretreatment provided highly significant protection against adenosine induced bronchoconstriction, with geometric mean PC20AMP values of 52.6 mg/ml (95% CI 35.2 to 78.7) after placebo and 123.9 mg/ml (95% CI 83.0 to 185.0) after montelukast (p=0.006). The geometric mean of the montelukast/placebo PC20AMP ratio was 2.4 (95% CI 1.3 to 4.2). Montelukast had no significant effect on 4 hour urinary excretion of LTE4 compared with placebo.
Conclusions: Selective CysLT1 receptor antagonism with montelukast provides highly significant protection against AMP induced bronchoconstriction in patients with atopic asthma, implying that cysteinyl leukotrienes are generated from airway mast cells through preferential activation of their A2B receptors.
- adenosine induced bronchoconstriction
- montelukast
- cysteinyl leukotrienes
- asthma
Statistics from Altmetric.com
Following the structural identification of slow reacting substance of anaphylaxis (SRS-A) as the three cysteinyl leukotrienes LTC4, LTD4, and LTE4, this mediator class has been a strong candidate for contributing to airways dysfunction in asthma. Their powerful bronchoconstrictor effect is mediated through the recently identified 7-transmembrane, G protein coupled cysteinyl leukotriene-1 (CysLT1) receptor that can be effectively antagonised by montelukast, zafirlukast, and pranlukast with mean IC50 values of 2.3 nM, 1.9 nM, and 4.3 nM, respectively.1 Cysteinyl leukotrienes contribute to the bronchoconstrictor actions of a number of bronchial provoking agents including allergen,2 exercise,3 cold air,4 sulphur dioxide,5 and aspirin,6 implicating their release as part of the indirect bronchoconstrictor response. The availability of selective antagonists acting at the CysLT1 receptor and inhibitors of 5-lipoxygenase (5-LO) that block the release of cysteinyl leukotrienes and LTB4 from mast cells, eosinophils, basophils, and macrophages has enabled the contribution of this mediator class to be assessed in a number of controlled laboratory challenges, including inhaled adenosine.7
Adenosine is an endogenous nucleoside released from metabolically active cells and generated extracellularly through the degradation of released adenosine nucleotides, including AMP, ADP, and ATP.8 It is a potent biological mediator that modulates the activity of numerous cell types including platelets, neutrophils, and mast cells via specific adenosine receptors (A1, A2A, A2B, A3).9 Among the many actions of adenosine, several lines of evidence suggest a contribution to the pathophysiology of asthma.10
Adenosine has aroused specific interest as a selective enhancer of mast cell mediator release acting through stimulation of adenosine A2B receptors on “primed” mast cells in the airways.8 A contribution of leukotrienes to adenosine induced bronchoconstriction in the human lung has been demonstrated in vitro.11 The only reported study of inhaled adenosine in asthma in vivo implicated leukotrienes using the experimental 5-LO inhibitor ABT-76112 which inhibits the synthesis both of cysteinyl leukotrienes and LTB4. We hypothesised that adenosine induced bronchoconstriction in asthma patients is specifically linked to the release of cysteinyl leukotrienes, and we report the first study in this model using a potent and selective antagonist of the CysLT1 receptor.
METHODS
Subjects
Eighteen subjects (seven men) of mean (SD) age 26.1 (8.4) years participated in the study (table 1). They were non-smokers with mild to moderate persistent asthma and were atopic as defined by positive skin prick tests (>3 mm wheal response) to one or more common aeroallergens (grass pollen, tree pollen, cat fur, dog hair, and Dermatophagoides pteronyssinus (Bayer Corporation, Elkhart, USA). Subjects had a baseline forced expiratory volume in 1 second (FEV1) ≥70% of their predicted values (or >1.5 l) and were only using short acting inhaled β2 adrenoreceptor agonists. None of the subjects was taking oral or inhaled corticosteroids or theophylline. Bronchodilators were withheld for 8 hours before each visit to the laboratory. No subject was studied within 4 weeks of an upper respiratory tract infection or exacerbation of asthma.
Characteristics of study patients
Subjects gave written informed consent and the study was approved by the Southampton and South West Hampshire local research ethics committee.
Study design
The study was of a randomised, double blind, placebo controlled, crossover design. With the sample size of 18, it was designed to have 80% power (p=0.05, two sided test) to detect a threefold increase in the provocative concentration of inhaled AMP needed to decrease FEV1 by 20% of baseline (PC20AMP). This was based on the PC20 being log10 normally distributed and within subject variance in log PC20 of 0.56.13
Each subject attended the laboratory on five occasions over a period of 4 weeks. At the first visit a medical and asthma history was taken, subjects underwent a full physical examination, and an electrocardiogram was performed. Before enrolment, a histamine bronchial challenge test was performed as a standard research screening procedure to establish baseline bronchial reactivity and therefore eligibility into the study. Subjects were included with a histamine PC20 of ≤ 16 mg/ml. Within 1 week of the screening visit they attended the department for an AMP inhalation bronchial provocation test, and were required to demonstrate a 20% fall in FEV1 after an AMP challenge to be eligible for the study. Urine was collected 4 hours after the AMP challenge. At the end of visit 2, subjects were randomly allocated the first 10 mg dose of montelukast (Singulair; Merck Sharp & Dohme Ltd, Hertfordshire, UK) or matched placebo orally. Visit 3 took place on the following day when the subject receiving the second dose of montelukast (10 mg) or placebo in the morning. An AMP challenge was then performed 4 hours after dosing and urine was collected 4 hours later. Serial FEV1 measurements were measured every 15 minutes for 1 hour after the AMP challenge. Subjects were crossed over after a washout period of 14 days and received either montelukast or placebo. All tests performed initially (AMP challenge, urinary LTE4 assay) were repeated.
Bronchial provocation
Pulmonary function was measured before and during the provocation using a dry wedge spirometer (Vitalograph Ltd, Buckinghamshire, UK). On each challenge day histamine acid phosphate (BCN Chemicals Inc, Beaconsfield, Quebec, Canada) and AMP (Sigma Chemical Co, Poole, Dorset, UK ) were made up freshly in 0.9% (w/v) sodium chloride solution to produce a range of increasing doubling concentrations from 0.03 to 32 mg/ml (0.1–104 mmol/l) and from 0.39 to 400 mg/ml (4.48–1151.6 mmol/l), respectively. The solutions were administered using a dosimeter (SPIRA Electro 2 inhalation dosimeter; Spira Resp Care Center Ltd, Helsinki, Finland). Wearing a nose clip, subjects inhaled the aerosolised solutions in five breaths from end tidal volume to full inspiratory capacity via a mouthpiece. Subjects were trained to take 3 seconds to reach full inspiratory capacity. All subjects were challenged using the same spirometer and dosimeter, which was calibrated before each patient use.
Baseline FEV1 was performed at the beginning of each challenge. After inhalation of 0.9% sodium chloride, repeated measurements of FEV1 at 1 and 3 minutes were noted, the higher value being recorded. If FEV1 did not fall by >10% of the baseline value, the bronchoprovocation challenge with histamine (screening) or AMP (study) was performed. Increasing doubling concentrations of the stimulant (histamine or AMP) were inhaled at 5 minute intervals until FEV1 had fallen by >20% of the post saline value or the highest concentration of the stimulant had been administered.
Urine collection for LTE4 quantification
Urine was collected in separate containers without preservatives 4 hours after the AMP challenge at visit 2 and 4 hours after the AMP challenge (8 hours after dosing) at visit 3. The same intervals were used during the crossover phase. Aliquots (30 ml) of each sample were stored at –20°C and assayed after a 5 month period. Urinary LTE4 levels were measured using a Biotrak leukotriene C4/D4/E4 enzyme immunoassay (EIA) system (Amersham Pharmacia Biotech UK Ltd, Amersham, Buckinghamshire, UK) as described and validated by Kumlin et al.14 Creatinine was measured in all urinary samples and the results expressed as pmol excreted LTE4/mmol creatinine.
Data analysis
Data were analysed using spss version 10 (SPSS, Chicago, IL, USA). The percentage decrease in FEV1 was plotted against the cumulative concentration of AMP inhaled on a logarithmic scale and the concentration of agonist producing a 20% fall from the post saline value (PC20) was determined by linear interpolation between the last two concentrations. Bronchoconstriction induced by AMP was expressed as concentration-response curves which were constructed as the logarithm of the concentration nebulised against the percentage change in FEV1. Least squares regression was performed on % FEV1 versus concentration, with treatment and concentration/treatment interaction terms. The interaction term was tested to assess parallelism of the response curves.
Both PC20 and urinary LTE4 levels were assumed to be log normally distributed. Analysis of covariance (ANCOVA), adjusting for subject, period and baseline, was used to estimate treatment effects on the log transformed data.15 The montelukast/placebo concentration ratio was calculated by dividing the PC20AMP value obtained after administration of montelukast by that obtained after placebo. The geometric mean and 95% confidence intervals of the montelukast/placebo concentration ratio were calculated by transforming back to the original measurement scale.
RESULTS
There were no significant differences in baseline or post saline FEV1 values on any of the study days. As part of the inclusion criteria, all subjects exhibited bronchial hyperreactivity to inhaled histamine with a baseline geometric mean PC20 histamine of 2.0 mg/ml (range 0.29–8.82 mg/ml; table 1). All patients in the study showed bronchial reactivity to AMP at baseline. From the patients enrolled, the FEV1 failed to fall by 20% after the placebo arm and after treatment with montelukast in one patient. This was different from this subject's baseline AMP challenge which showed a PC20AMP. This difference compared with baseline can probably be explained by the expected variability of the bronchial challenge procedure. Two subjects failed to achieve a PC20 after montelukast treatment. This differed from their baseline AMP challenge where a PC20 was recorded, illustrating the protective effect of montelukast in some patients. Subjects who failed to show a 20% fall in FEV1 were assigned a PC20 of 1600 mg/ml (twice the maximum cumulative concentration).
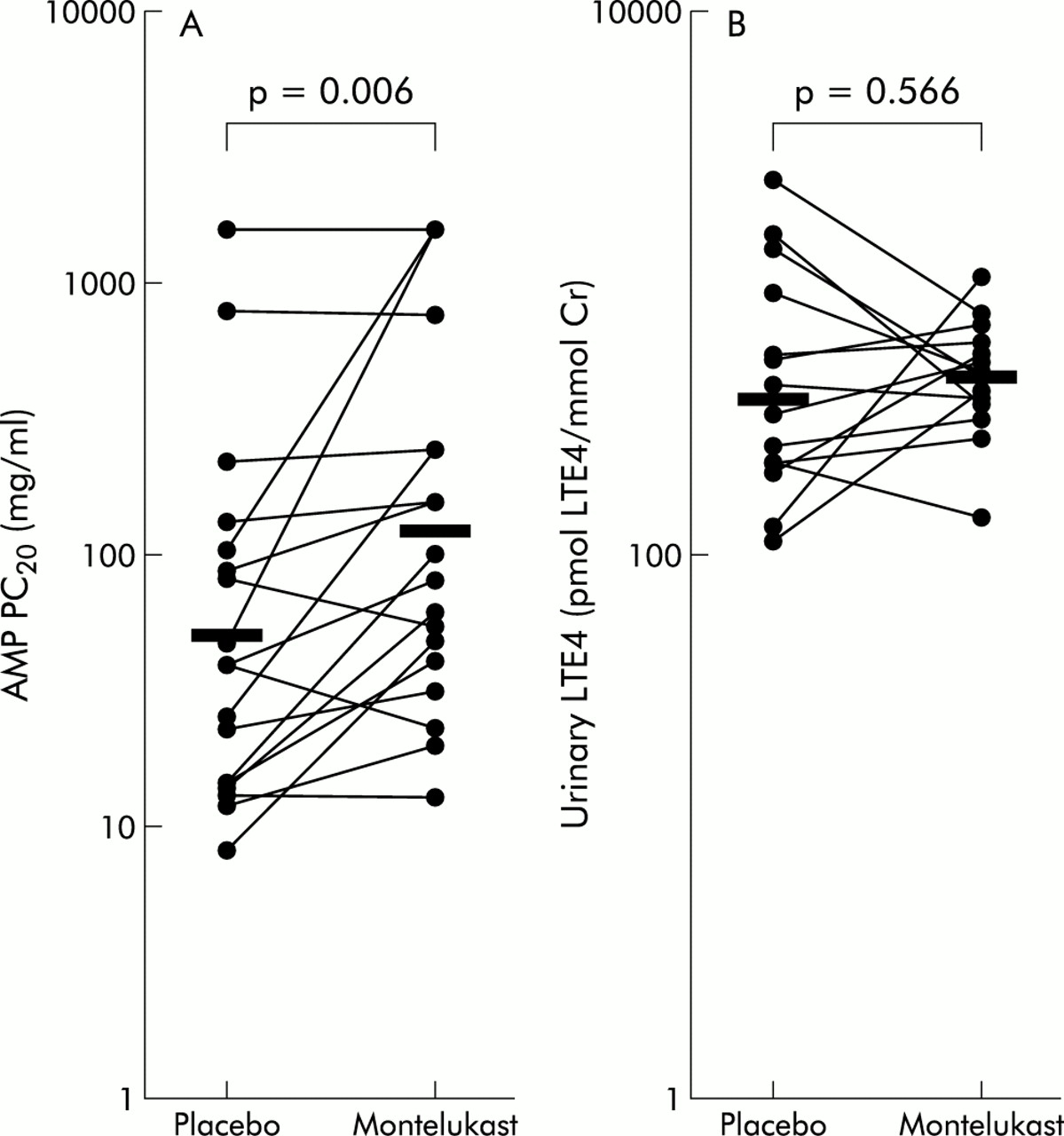
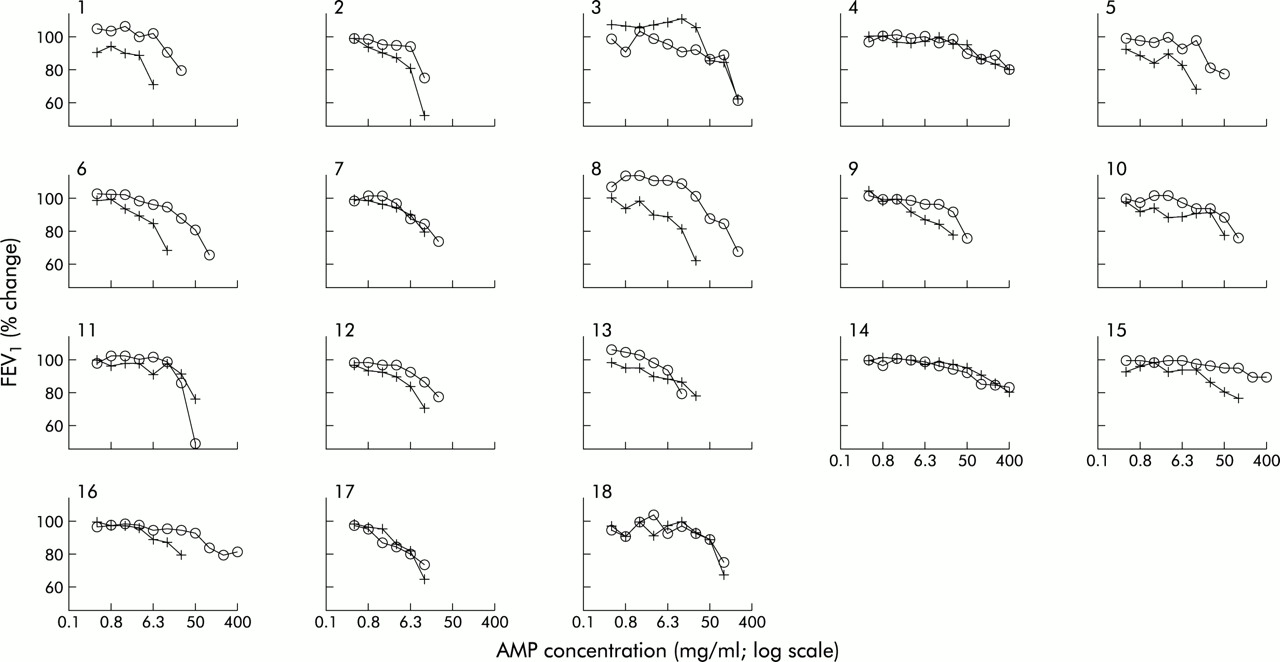
The slopes of the concentration-response curves did not depart significantly from parallel (p>0.05). There was a displacement to the right of the AMP concentration-response curve in 13 of the 18 subjects (fig 1). The geometric mean (range) concentration of AMP required to produce a 20% decrease in FEV1 PC20 (PC20AMP) was 51.6 mg/ml (range 8.42–1600) after placebo and 126.5 mg/ml (range 13.31–1600) after montelukast (fig 2A). Thirteen subjects had an increased PC20AMP after montelukast (montelukast/placebo concentration ratio >1) while five subjects had little or no change in PC20AMP. Adjusting for period and baseline effects, the treatment effect of montelukast remained statistically significant (p=0.006). The estimated geometric mean and 95% confidence interval for placebo and treatment were 52.6 mg/ml (35.2 to 78.7) and 123.9 mg/ml (83.0 to 185.0), respectively. The geometric mean of the montelukast/placebo concentration ratio was 2.4 (95% CI 1.3 to 4.2). The analysis was repeated omitting the three subjects who failed to show a decrease in FEV1 of at least 20% from the post-saline FEV1 after treatment with placebo or montelukast. Using ANCOVA, the treatment effect of montelukast remained statistically significant (p=0.004), with an estimated geometric mean of 42.7 mg/ml (95% CI 32.8 to 55.5) and 82.0 mg/ml (95% CI 60.4 to 111.2) for placebo and montelukast, respectively. The geometric mean of the montelukast/placebo concentration ratio was 1.9 (95% CI 1.3 to 2.9).
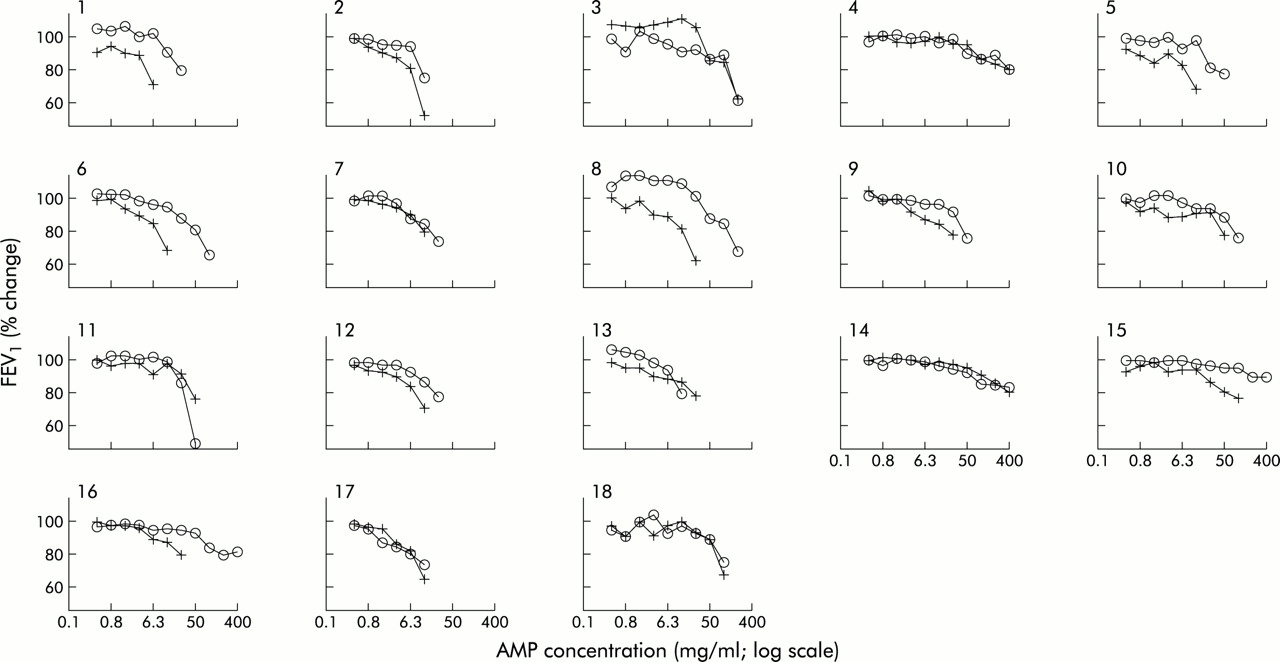
Effects of oral placebo (+) and 10 mg montelukast (∘) on AMP induced falls in FEV1 in 18 patients with atopic asthma.
{kind=link}
{kind=link}
Effects of oral placebo and 10 mg montelukast on (A) bronchoconstriction provoked by AMP in 18 patients with atopic asthma and (B) urinary LTE4 levels 4 hours after an AMP challenge. The horizontal bars denote geometric mean values.
Complete data for evaluation of urinary LTE4 levels were available in 14 of the 18 subjects. The geometric mean urinary LTE4 level was 210.4 pmol/mmol creatinine (95% CI 168.9 to 268.4) after placebo and 230.9 pmol/mmol creatinine (95% CI 201.7 to 264.4) after montelukast (fig 2B).
The ratio of urinary LTE4 levels after montelukast to after placebo was 0.9 (95% CI 0.7 to 1.2), p=0.566 (ANCOVA).
DISCUSSION
Consistent with previous studies, we have shown that inhaled AMP induces bronchoconstriction in patients with asthma.16,17 We have further shown that the selective CysLT1 receptor antagonist montelukast is a potent but partial antagonist of AMP induced bronchoconstriction, and that montelukast was not associated with a significant reduction in the mean post challenge urinary LTE4 levels. The ability of montelukast to inhibit adenosine induced bronchoconstriction through CysLT1 receptor blockade thus definitively establishes that cysteinyl leukotrienes contribute as mediators of adenosine induced bronchoconstriction.
Although the exact mechanism of bronchoconstriction produced by inhaled adenosine in asthma is not known, previous studies in vitro have shown that adenosine potentiates the release of both preformed and newly synthesised mediators of inflammation from human lung mast cells18 through stimulation of A2B receptors and activation of the phosphatidylinositol cascade.8,19 The contribution of histamine has been shown using the selective histamine H1 receptor antagonists terfenadine, astemizole, and loratadine,20 and strongly supports the idea that inhaled AMP potentiates the release of preformed histamine from immunologically primed mast cells in asthmatic airways.21
The contribution of cysteinyl leukotrienes to bronchoconstrictor responses of AMP is supported by the inhibitory effects of the cysteinyl leukotriene receptor antagonist ICI 198,615 and the FLAP (five lipoxygenase activating protein) inhibitor MK-886 against adenosine provoked contractile responses of asthmatic airways in vitro,11 which suggests that adenosine acts indirectly by liberation of leukotrienes possibly from mast cells. A contribution of eicosanoids in vivo is indicated by the attenuation of adenosine induced bronchoconstriction in patients treated with selective inhibitors of cyclooxygenase22 and the experimental 5-LO inhibitor ABT-761.12 Eicosanoids generated by human lung mast cells include PGD2, LTB4, and cysteinyl leukotrienes. The availability of potent and selective CysLT1 antagonists has enabled us to define for the first time the specific contribution of cysteinyl leukotrienes to bronchoconstriction provoked by AMP.
In the present study montelukast produced a 2.9 fold increase in the concentration of AMP required for an equivalent fall in FEV1 after placebo. Three of the 18 subjects failed to show a decrease of 20% in their FEV1 from the post saline value after the maximum concentration of AMP was administered. Although a censored value of 1600 mg/ml was assigned to these subjects, when omitted from the analysis the treatment effect of montelukast still revealed a twofold protection against the bronchoconstrictor effect of AMP which remained highly significant (p=0.004).
The magnitude of the protection observed in vivo is considerably less than the 16-fold protection afforded by histamine H1 receptor antagonism.23 This difference in the relative inhibitory capacity of antagonistic inhibitors emphasises the central role that histamine plays in adenosine induced bronchoconstriction and emphasises that, while augmenting mast cell mediator release, stimulation of A2B receptors preferentially augments degranulation with release of preformed mediators rather than stimulating the production of newly generated mediators.21 Although histamine would seem to be central to the adenosine induced bronchoconstriction response in most of our subjects, three demonstrated a predominantly leukotriene driven adenosine response as illustrated by a failure to achieve a PC20 after the maximum concentration of AMP (400 mg/ml) was inhaled following montelukast (fig 1). This adds to the view that, within the asthmatic population, there is substantial heterogeneity among individuals in their therapeutic response to drugs that selectively interfere with mediator pathways. The reason(s) for such heterogeneity in the relative contribution of any one mediator to bronchoconstriction produced by an indirect stimulus is not known. In the case of leukotrienes, one possibility is promoter polymorphism as indicated in the 5-LO24 and leukotriene C4 synthase genes.25
Urinary excretion of LTE4 has often been used as a marker of cysteinyl leukotriene production in the airways. LTE4 concentrations in urine are significantly increased in response to allergen challenge,26 aspirin challenge in aspirin sensitive asthmatic subjects,27 and during spontaneous exacerbations of asthma.26 In the present study pretreatment with montelukast was not associated with a significant reduction in the mean post challenge urinary LTE4 levels when compared with placebo, in contrast to the effects of the 5-LO inhibitor ABT-761,12 consistent with the different modes of action of these drugs.
In conclusion, our results confirm that selective CysLT1 receptor antagonism produces a small but highly significant protection against AMP induced bronchoconstriction in subjects with atopic asthma, indicating that cysteinyl leukotrienes are generated by this indirect challenge probably from airway mast cells through preferential activation of their A2B receptors. While the contribution of cysteinyl leukotrienes to the adenosine induced bronchoconstriction response is likely to be smaller than that of histamine overall, we have shown that cysteinyl leukotrienes play a markedly more pronounced role in a minority of subjects. These findings constitute the basis for further clinical studies to investigate whether the adenosine bronchoconstrictor response can be completely attenuated by a combination of histamine H1 receptor and selective CysLT1 receptor blockade as described in vitro.11
REFERENCES
Footnotes
-
This study was funded by Merck Sharp & Dohme Ltd, Herts, UK.