Article Text

Statistics from Altmetric.com
Chronic obstructive pulmonary disease (COPD) is the fifth leading cause of death in the UK,1 causing 26 000 deaths and 240 000 hospital admissions and costing the NHS £486 million per annum. It is also a global problem, reaching almost epidemic proportions in the developing world. Despite this, it receives relatively little attention compared with diseases with a similar impact such as coronary heart disease and cancer. The research into new treatments to prevent the development and progression of this condition presents a major challenge. It is thus important to develop tools that can both direct development of new drugs and provide insights into assessment of their efficacy before extensive human trials are undertaken.
Animal models act as a bridge between in vitro studies in the laboratory and studies in humans. As such, they have had a major impact on the investigation of many medical conditions. In COPD, in particular, animal models would enable basic research to investigate the mechanisms of inflammatory cell recruitment and abnormal matrix repair (the proteinase/antiproteinase hypothesis2) and alternative hypotheses of the pathogenesis of emphysema such as those implicating lung cell apoptosis as a primary event.3 They would also facilitate the testing of new treatments such as gene therapy (in cystic fibrosis and α1-antitrypsin deficiency (A1AT)) and “designer drugs” targeting specific cytokines or biochemical pathways. By leading to a clearer understanding of the key events in the pathophysiology of COPD and enabling short term studies to develop appropriate strategies, animal models can provide a framework for the rational and safe design of expensive and long term clinical studies.
Advantages and disadvantages of mouse models
The mouse provides the best choice for an animal model4 because the mouse genome has been extensively studied and sequenced and close similarities exist with the human genome. In addition, complementary antibodies and probes exist to many mouse enzymes, enabling them to be studied directly for quantification and localisation. There is also a rapid reproductive turnover, with large litter sizes and a short lifespan enabling studies to be completed within months. Breeding, housing, and maintenance costs are relatively low.
However, animal models have a number of limitations that have to be borne in mind. There are certain anatomical and physiological differences between the respiratory tract of mice and humans.5 For instance, in mice there are no extensive cilia, few submucosal glands in the trachea, and no goblet cells. Mice do not expectorate sputum, are obligate nasal breathers that filter tobacco smoke inefficiently, and have less branching of the bronchial tree without respiratory bronchioles. The profile of inflammatory mediators is also slightly different in the mouse. For instance, the interstitial collagenase MMP-1 is found in humans but not mice6 and, although mice have receptors for important pro-inflammatory cytokines found in humans such as interleukin (IL)-87 and leukotriene B (LTB)-4,8 ,9 their role in mice has not been ascertained. Extrapolations of findings from the mouse to the human therefore have to be made with these differences in mind.
First animal models: exposures
The first animal models of COPD stemmed from the extrinsic exposure to inflammatory stresses. Gross et alin 1965 described the first reproducible model of emphysema by instilling papain (a plant protease) into the lungs of rats.10 Together with the observation of Laurell and Eriksson two years earlier of an association between serum A1AT (a protease inhibitor) deficiency and chronic airways disease and emphysema,11 this animal model provided the basis of the proteinase-antiproteinase hypothesis of emphysema. Various proteases have since been introduced into the lungs of laboratory animals. However, these are relatively crude models in which the lung injury is caused by a single massive insult rather than a continuous low grade inflammatory process (which is believed to underlie smoking related emphysema). Also, the delivery of proteinases into the airways by a bolus in solution may not reflect the processes resulting from delivery of the enzymes in quantum packets by viable inflammatory cells to the interstitium in human disease.12 Nevertheless, such models can be useful for studying events after the insult, as in the study of alveolar repair in response to treatment with a retinoic acid derivative13 following instillation of porcine pancreatic elastase to create the initial emphysema model in rats.
There are several tobacco smoking animal models of emphysema. Certain laboratory strains of mice are more susceptible to the effects of cigarette smoke—for example, C57BL/6, fig 1—and rats are less susceptible than mice. Dhami et al 14 used C57BL/6 mice exposed to the smoke of two cigarettes using a standard smoking apparatus to demonstrate that the administration of human A1AT or antineutrophil antibodies prevented the early breakdown of elastin and collagen, supporting the role of neutrophil elastase in acute lung damage due to cigarette smoke. However, in other smoking models the accumulation of macrophages has also been shown to be a feature necessary for producing some of the long term pathological features of emphysema.15

Effect of cigarette smoke exposure on airspaces of C57BL/6 mice. Scanning electron microscopy ×400 of (A) lungs of mice exposed to cigarette smoke for 6 months and (B) lungs of non-exposed age-matched controls. Reprinted from Shapiro with permission of the publishers.27
In order to create a model of A1AT deficiency,d-galactosamine has been administered intraperitoneally to rats,16 resulting in decreased circulating A1AT levels and a propensity for more severe emphysema after the intravenous injection of pancreatic elastase. The emphysema was argued to have been caused by a proteinase/antiproteinase imbalance, but other glycoproteins that are also affected by d-galactosamine may have influenced the development of emphysema.
Models resulting from natural mutations
Some naturally occurring mutant strains of C57BL/6 mice are more prone to developing emphysema including tight skin, pallid andblotchy variants. They tend to have lower serum concentrations of A1AT but this alone is not thought to be responsible for the development of emphysema. The airspace enlargement probably results from abnormalities in lung development rather than destruction of mature lung tissue. Tight skin mice17 have a mutation of the fibrillin-1 gene that affects formation of elastic fibres which are central to normal alveolar development. Pallidmice18 have a mutation affecting syntaxin 1319 (a cell membrane protein) that gives rise to gradual development and progression of emphysema.Blotchy mice20 have abnormal translation of the Menkes gene on the X chromosome and develop a connective tissue disorder similar to the human cutis laxa with associated abnormal lung matrix. These models may provide further insight into the importance and dynamics of normal lung matrix formation, damage, and repair. Their role in determining the pathogenesis and treatment of human emphysema remains unknown, although cutis laxa is associated with the development of emphysema in man.21
“Knockout” mouse models
“Knockout” mouse models have been developed to disrupt the expression of a targeted gene as a result of the insertion of another gene or nucleotide sequence, hence they are often called “loss of function” models.
They are created by the process of targeted mutagenesis.22This relies on a “targeting” construct that has flanking sequences that match those in the gene of interest and contains markers for positive and negative selection, such as the phosphoglycerate kinase/bacterial neomycin resistance gene (PGK-neo) and phosphoglycerate kinase/viral thymidine kinase gene (PGK-tk), respectively. The construct is microinjected into embryonic stem (ES) cells obtained from the inner cell mass of a blastocyst, which have the potential to differentiate into any tissue in the body. “Homologous recombination” then occurs resulting in exchange of the artificial DNA sequence for the corresponding area of genomic DNA as the DNA breaks and rejoins. The probability of this happening is increased by the presence of the matching sequences that flank the target gene. The ES cells (from black mice) that have correctly recombined with the construct are selected out using neomycin and ganciclovir (due to the presence of the PGK-neo gene and absence of the PGK-tk gene). These are then injected into the host mouse blastocyst (from white mice) and impregnated into a foster mother. The result is a chimeric mouse (black and white). Chimeras that incorporate the ES cells in their germ line (for instance, the sperm), when bred with white mice will produce offspring heterozygous for the gene deletion (speckled or “Agouti” mice), and interbreeding of these in turn will lead to homozygous offspring (black mice) for the required mutation. The targeted mutation can thus be followed by coat colour (fig 2).
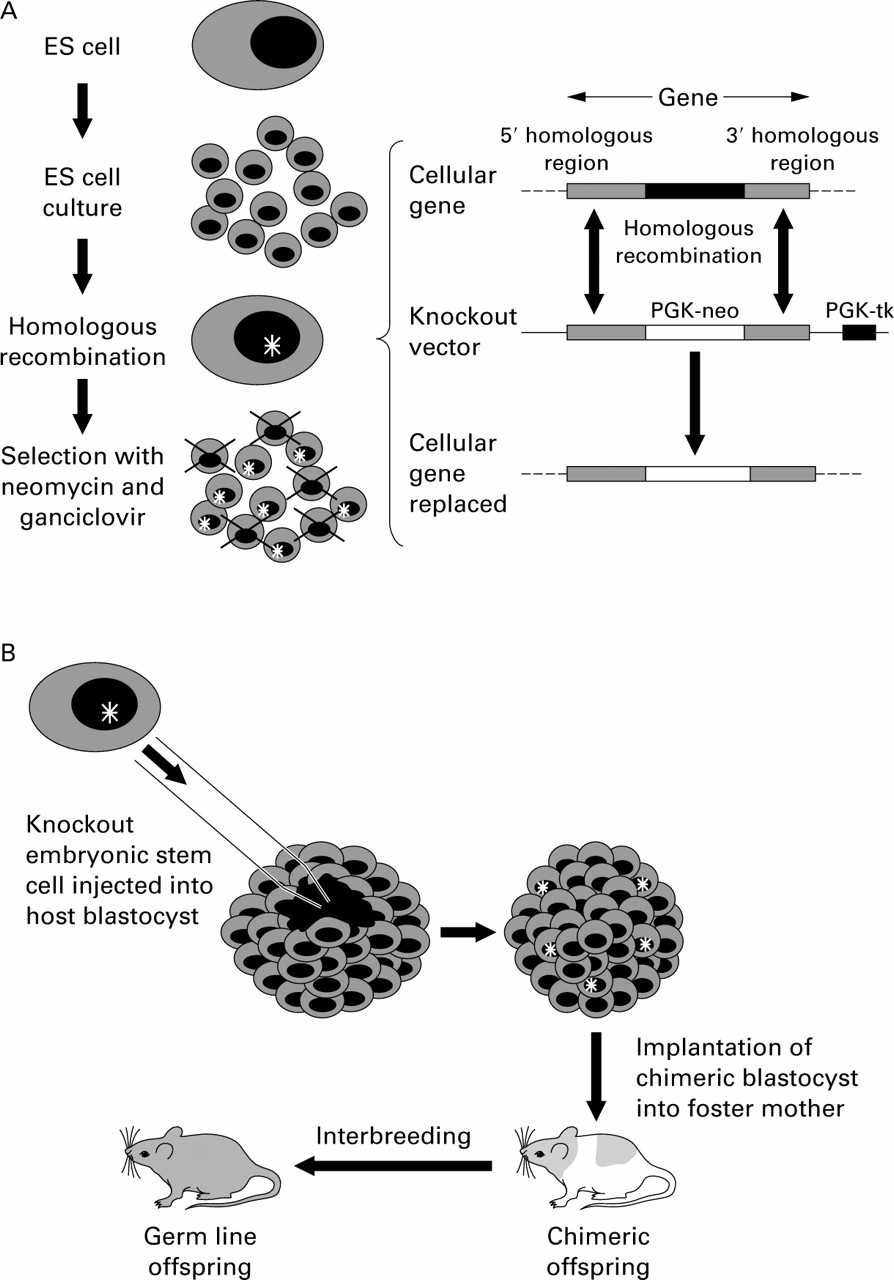
Method of targeted mutagenesis (based on Majzoub and Muglia22). (A) A knockout vector is created consisting of the phosphoglycerate kinase/bacterial neomycin resistance gene (PGK-neo) flanked by segments homologous to the cellular gene and the phosphoglycerate kinase/viral thymidine kinase gene (PGK-tk) downstream. The vector is introduced into embryonic stem (ES) cell culture from black mice. Homologous recombination occurs between the vector and the cellular gene at the matching flanking regions, resulting in incorporation of the “knockout” vector, including the PGK-neo but not the PGK-tk, into the genome of the ES cell. The presence of the PGK-neo and the absence of PGK-tk allow survival of these ES cells after positive/negative selection with neomycin and ganciclovir. (B) The mutant clone of ES cells is then microinjected into the host blastocyst (from a white mouse) which is implanted into a foster mother resulting in the development of a chimeric mouse. The incorporation of the ES cells into the germ line of the chimeric mouse results in heterozygous germ line transmission of the mutant gene. The heterozygotes are then mated to produce offspring homozygous for the mutant gene, identified by reversion to the black phenotype.
This strategy, if successful, results in mice that develop in utero with a single gene deletion. The major problem with this approach is that the gene defect may also affect prenatal and postnatal development leading to changes that may be relevant to disease expression in later life. Alternatively, there may be a degree of overlapping function in metabolic processes so that normal development is protected against the loss of the gene (redundancy). However, absence of one gene during development may lead to upregulation of components in compensatory pathways, thereby complicating the final phenotype of the “knockout” mouse.
Ideally, therefore, “knockout” animals should be allowed to develop normally before the gene of interest is deleted. This can be achieved with the Cre-lox P technology23 which involves the insertion of specific nucleotide sequences called lox P either side of the target gene. The Cre enzyme (Cre recombinase, derived from a bacteriophage P1 that infects Escherichia coli) recognises the sequences flanked by the lox P sequences and causes DNA cleavage at these points, thereby deleting the gene. The advantage of this system is that relatively large sections of DNA can be excised after transfection of ES cells with Cre. Also, the Cre gene itself can be inserted into the mouse genome and modified so that it can be expressed only in specific tissue by being linked to a cell specific promoter. When this mouse is crossed with a mouse that has a target gene flanked by lox P sequences, this gene will only be “knocked out” in those specific tissue cells. The Cre gene can also be made to be inducible by using a cell specific promoter that can be activated by the presence of another molecule. For instance, promoter Mx1 in T cells can be induced by interferon (IFN)α leading to the expression of the linked DNA-polymerase-β gene.24 Gene deletion can therefore be controlled in time and place by administration of the inducing agent (such as IFNα) to the mice leading to the controlled expression of Cre and hence cleavage of the target gene again only in the cells of interest. These sophisticated techniques remove the complicating issues of possible interference with developmental processes by “knocking out” the gene after development has occurred, and lack of tissue specificity by only deleting the product in cells of interest (fig 3).

Strategies for creating “knockout” mice (after Shapiro5). (A) The standard knockout is formed from the incorporation of the neomycin resistance gene driven by the phosphoglycerate promoter (PGK-neo) into gene X. This mutation introduced into embryonic stem cells renders this gene inactive throughout development and adult life. (B) Conditional knockouts achieve cell specific gene deletion by flanking gene X with lox P sequences. When these mice are crossed with a transgenic mouse expressing Cre recombinase under cell specific control, the gene X is only excised in those cells expressing Cre. (C) Inducible knockouts use the same strategy as conditional knockouts but the Cre gene is driven by a promoter that only allows transcription in the presence of the inducing agent. In this example the interferon (IFN)α inducible promoter Mx1 is shown.22 IFNα can be introduced when gene knockout is specifically required.
“Knockout” models for emphysema include those for platelet derived growth factor A25 which lose myofibroblasts and therefore have decreased elastin deposition, and the double “knockout” mutants for fibroblast growth factor receptors 3 and 426which exhibit abnormal alveolar formation and septation. It is also noteworthy that both macrophage elastase15 and neutrophil elastase “knockout” mice27 are protected from tobacco smoke induced emphysema, supporting the role of these genes in the pathogenesis of emphysema.
Cystic fibrosis is caused by a single gene defect and is a common disease affecting a young population. As a result of an extensive research effort, several “knockout” mouse models for this condition exist.28-31 These have been important in the study of the mechanisms of lung and intestinal disease in this condition, as well as potentially providing a system for the development and testing of treatments for patients with cystic fibrosis.
Transgenic mouse models
Transgenic mice have genes of interest inserted into their genome with the resultant expression or overexpression of the relevant product, and hence are often called “gain of function” models. Various transgenes have given rise to the development of emphysema in mice, often discovered serendipitously. The metalloproteinase-1 (interstitial collagenase) transgene linked with the haptoglobin promoter unexpectedly caused emphysema as a result of expression in the lung.32 Again, whether this was due to an effect on lung development or due to destruction of mature lung connective tissue is unknown. Transgenic mice overexpressing platelet-derived growth factor B, driven by the lung specific surfactant protein C promoter, also developed emphysema,33 but this was in combination with fibrosis and therefore may have been due to tethering of the airways which is a process analogous to the focal emphysema of coal workers' pneumoconiosis.
The klotho mouse34 was originally intended to be a model for hypertension as a consequence of the insertion of a sodium channel transgene leading to hypernatraemia. This primary aim failed, but the animals were observed to exhibit signs of premature ageing and airspace enlargement. It was found that the transgene had disrupted the klotho gene which codes for a transmembrane protein. It is uncertain whether the lungs ever develop fully in these mice before emphysema occurs, so its role in the study of the human ageing process in the lungs is uncertain.
The disadvantage with transgenic methods in elucidating the role of a gene product in the aetiology of adult emphysema is that the gene of interest is also expressed throughout organ development and growth, rather than at the preferred time after maturation. This problem led to the development of the technique of inducible transgenic expression. Zheng and colleagues35 targeted overexpression of IL-13—a T helper 2 (Th2) pathway cytokine usually associated with atopy and asthma—to the lungs of mice. The authors used two constructs that included the Clara cell promoter CC10 which targets expression to the lung, and the reverse tetracycline transactivator (rtTA) which allows IL-13 gene transcription only in the presence of tetracycline given in the diet when required. They found that these mice develop emphysema, mucous metaplasia, and inflammation which are metalloproteinase and cysteine proteinase dependent. Similar but more gradual pathological changes occurred using the same method of inducible overexpression of IFNγ,36 a T helper 1 (Th1) pathway cytokine associated with inflammatory reaction to infection in the lungs. These two models suggest that the pathogenesis of emphysema is complex, implicating both Th1 and Th2 pathways.
The question of development dependent and independent phenotypic expression has been addressed by the same CC10-rtTA method in a study on the effect of a transgene for IL-11 which caused emphysema in mice when constitutively expressed but not when inducibly expressed after lung development.37 The results suggested that IL-11 exerts its effect during lung development, although the mechanism remains unknown.
The next generation: “knock out”/”knock in” mice
Following the “knock out” of murine genes, the technology is being developed to insert (or “knock in”) equivalent human genes in their place. In relation to pulmonary medicine, this is of major relevance in A1AT deficiency where the human variants (M, Z and S) can be inserted into the mouse genome after the murine A1AT gene has been “knocked out”.
In mice there is known to be a cluster of four or five A1AT-like genes, depending on the strain, on chromosome 12 (S Shapiro, N Kalsheker, personal communication). This is different from humans where there is just one functional gene38 and one pseudogene of unknown importance39 on chromosome 14. Elucidation of the base sequence of the A1AT gene cluster in the mouse followed by the design of specific constructs will enable the “knock out” of the mouse gene locus and the subsequent “knock in” of the human equivalent. This will generate a more accurate model of A1AT deficiency than the straightforward “knockout” (which is the equivalent of a null mutation) since the mutant human genes will be expressed and the abnormal human A1AT protein will be produced and hence “tolerated” during development.
The model could then be used to research the pathogenesis of emphysema and bronchial disease in A1AT deficiency more accurately, since it should represent the human equivalent of reduced secretion of the abnormal protein and intracellular accumulation due to polymerisation.40 The mice could then be exposed to a variety of relevant inflammatory stimuli—including cigarette smoke, elastase, cytokines, infections or endotoxin—to study their relevance in the development of human disease. In addition, the mice could be used to test treatments such as A1AT augmentation since repeated doses of human A1AT could be given without the development of an immune response. The standard “knockout” mice are likely to react to the “foreign” human epitopes on the second and subsequent administrations, leading to the development of antibodies that prevent the efficacy of repeated treatments. These models could also be used to test the efficacy of gene therapies with adenoviral41 or adenovirus associated42 vectors that deliver the genetic information for producing protective proteins such as A1AT to the lung. Other treatments that could be tested using these models include peptides to block intracellular A1AT polymerisation of the abnormal Z protein,40 drugs that increase cellular secretion of A1AT,43 and drugs that inhibit elastase production or secretion.44
It is possible to develop further models by crossing A1AT deficient mice with other human A1AT variants to study the effect (if any) of the heterozygous state. In addition, cross breeding with neutrophil elastase “knockout” mice or neutrophil deficient mice will lead to further understanding of the interrelationship of these key antagonistic elements in the development of disease. It would enable the proteinase/antiproteinase hypothesis to be confirmed if the double mutants of the key pathogenetic components revert to the same susceptibility to emphysema as the wild-type mice.
Conclusion
Rodents and humans have certain anatomical and physiological differences in their bronchopulmonary trees but mount similar host responses to lung challenge. Animal models of COPD have evolved from the rather crude one-hit elastase exposure models to the sophisticated mutant strains being created by techniques at the cutting edge of genetic science. These advances enable us to enter a new era of scientific research into COPD, where individual mediators and their abrogation can be studied specifically and in detail. Such approaches will not only increase our understanding about pathogenesis, but will also facilitate the development and introduction of new therapeutic strategies.

{kind=link}
{kind=link}
{kind=link}
{kind=link}